A l'interface entre neurodégénérescence et oncologie
De multiples causes peuvent amener la genèse de maladies neurodégénératives primaires et de facon similaire nombre de maladies se manifestent par des symtomes cliniques identiques. Parmi les plus complexes figure le mimétisme de la sclérose latérale amyotrophique (SLA) par les syndromes neurologiques paranéoplasiques (SNP). Si la SLA est traditionnellement considérée comme une maladie idiopathique d'origine protéopathique (des aggrégats de protéines mal formées, mal localisées), des articles récurrents suggèrent qu'un sous-groupe de cas de syndromes du motoneurone (SMN) serait en réalité un épiphénomène d'une affection maligne sous-jacente.
Physiopathologie des syndromes paranéoplasiques
Un syndrome paranéoplasique se définit par des manifestations cliniques systémiques survenant à distance d'une tumeur primitive ou de ses métastases. Contrairement à l’effet de masse direct d’une tumeur, ces syndromes sont induits par deux mécanismes principaux, la production ectopique d’hormones, de cytokines ou de peptides par les cellules tumorales (par exemple, production d’ACTH dans le cancer du poumon à petites cellules) et l'expression par une tumeur d’antigènes onconeuronaux, normalement localisés dans le système nerveux. Le système immunitaire, cherchant à éradiquer la tumeur, produit des anticorps et des réponses des lymphocytes T qui traversent la barrière hémato-encéphalique et ciblent les neurones sains. Ceux-ci se détériorent et provoquent les symptomes de la SLA.
Relation entre l’oncogenèse et la neurodégénérescence
La relation entre l’oncogenèse et la neurodégénérescence motrice est caractérisée par un paradoxe statistique complexe. Les données épidémiologiques suggèrent fréquemment une corrélation inverse entre la survenue d’un cancer et celle de la SLA. Les patients atteints de SLA semblent présenter une incidence de cancer inférieure à la moyenne, et inversement. Ceci a conduit à l'hypothèse que ces deux affections représentent les extrémités opposées d'un spectre biologique : le cancer étant un état de prolifération cellulaire incontrôlée (échappant à l'apoptose), et la SLA un état de mort cellulaire prématurée (apoptose accélérée).
Cependant, certaines architectures génétiques complexifient cette dichotomie. Par exemple, des mutations du gène FUS (Fused in Sarcoma) – une cause connue de SLA familiale – sont également associées à certains liposarcomes. Dans ces cas, la même protéinopathie qui entraîne la mort des motoneurones peut aussi jouer un rôle dans la transformation oncogénique, suggérant que la « relation inverse » n'est pas universelle et dépend fortement de la voie moléculaire spécifique impliquée.
La SLA peut-elle être liée à une manifestation paranéoplasique?
Dans une étude publiée dans Brain Communications, Koropouli et ses collègues ont réalisé une méta-analyse des données individuelles de 163 patients afin de lever l'ambiguïté entourant le syndrome paranéoplasique de la SLA. Leurs résultats constituent une preuve de concept que la SLA peut effectivement être une manifestation paranéoplasique légitime.
Bien qu'habituellement j'accorde peu de crédits aux méta-analyses, voici les principaux enseignements de l'étude :
La similitude phénotypique : La SLA paranéoplasique peut se manifester sous forme de SLA, de sclérose latérale primitive (SLP) ou d'amyotrophie musculaire progressive (AMP), la rendant cliniquement indiscernable des formes idiopathiques sur la seule base de l'examen physique.
Le précédent temporel : Dans la majorité des cas, les symptômes neurologiques ont précédé le diagnostic de cancer. Ceci identifie la SLA comme un symptôme « sentinelle » potentiel de tumeurs malignes occultes, en particulier au niveau des poumons, des seins ou du système lymphatique.
Une vitesse d'évolution caractéristique : L'indicateur clinique le plus fiable d'une origine paranéoplasique était la progression rapide des déficits moteurs par rapport à l'évolution typique de la SLA idiopathique.
Des marqueurs immunologiques : Un sous-groupe significatif de patients présentait des marqueurs inflammatoires dans le liquide céphalo-rachidien (LCR), tels qu'une pléiocytose ou des bandes oligoclonales, ainsi que des anticorps onconeuronaux à haut risque comme SOX1.
Un impératif clinique : Le rôle des biomarqueurs néocancéreux dans le diagnostic de la SLA
L'importance majeure de l'identification d'une origine paranéoplasique aux symptômes de la maladie du motoneurone réside dans la possibilité de réversibilité ou de stabilisation. Alors que la SLA idiopathique demeure terminale et largement réfractaire au traitement, la maladie du motoneurone paranéoplasique peut répondre à une thérapie antinéoplasique intensive ou à une immunothérapie.
Par conséquent, l'étude souligne qu'une forte suspicion de syndrome paranéoplasique doit déclencher un bilan diagnostique rigoureux avant de poser le diagnostic définitif de SLA. En intégrant les biomarqueurs du néocancer dans l'algorithme de diagnostic, les cliniciens peuvent aller au-delà du phénotypage purement descriptif et s'attaquer aux facteurs immunitaires sous-jacents potentiels de la maladie, garantissant ainsi que les imitations traitables ne soient pas négligées dans l'ombre d'un diagnostic de SLA.
 By testing this approach on mouse models of both diseases and on human patient stem cells (iPSCs), the team observed several crucial improvements:
By testing this approach on mouse models of both diseases and on human patient stem cells (iPSCs), the team observed several crucial improvements: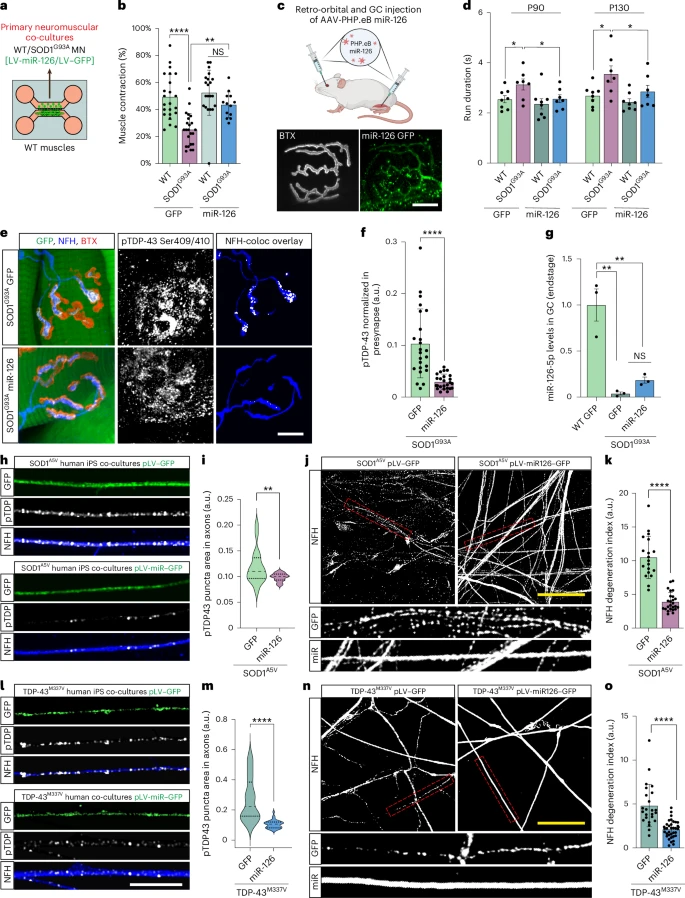 When pathologists examine the spinal motor neurons of patients with SOD1-related ALS, the nuclei generally appear normal: the TDP-43 protein is always present, and abnormal aggregates are rarely observed. This is why SOD1-related ALS has been considered "TDP-43 negative."
When pathologists examine the spinal motor neurons of patients with SOD1-related ALS, the nuclei generally appear normal: the TDP-43 protein is always present, and abnormal aggregates are rarely observed. This is why SOD1-related ALS has been considered "TDP-43 negative." Analyses statistiques contestables
Analyses statistiques contestables