L’hypothermie thérapeutique est maintenant la norme de soins pour différentes conditions impliquant la mort cellulaire.
Les mécanismes d'action de l'hypothermie thérapeutique sont variés et mal compris. Ils comprennent des mécanismes dont on pense qu'ils sont aussi impliqués dans la SLA.
Pour autant la possibilité que l'hypothermie thérapeutique a une application à la SLA n'avait pas été examinée.
Lee J Martin, Mark V Niedzwiecki et Margaret Wong, les auteurs de l’article examiné aujourd’hui, ont donc testé dans un modèle murin transgénique de la SLA, l'hypothèse selon laquelle l'hypothermie légère chronique intermittente a une efficacité thérapeutique.
Les souris ont été observées deux fois puis 4 à 5 fois par jour au stade terminal de la maladie.
Les modèles de souris SOD1 sont chroniquement fébriles. À 6 semaines d'âge, la température interne des modèle de souris SOD1 était significativement plus élevé (38 °C) par rapport aux souris témoins non transgéniques (37 °C).
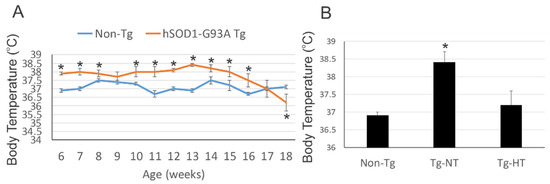
À 8 semaines d'âge, les souris SOD1 ont été randomisés dans plusieurs groupes. Le modèle de souris SOD1 avait une température corporelle inférieure de 1 à 2 ° C par rapport au modèle de souris SOD1 ne subissant pas de refroidissement.
Les températures corporelles de prérefroidissement parmi les groupes de modèles de souris SOD1 ne différaient pas de manière significative. Le réchauffement de la souris était lent et spontané à température ambiante. A température ambiante, l'activité motrice a été testée sur une roue de course à activité volontaire.
L'apparition de la maladie a été évaluée quantitativement par un déficit d'activité et de manière descriptive par la parésie des membres postérieurs. Pour évaluer l'efficacité de l'hypothermie, un groupe de souris a été euthanasié avant le stade final à l'âge de 12 semaines.
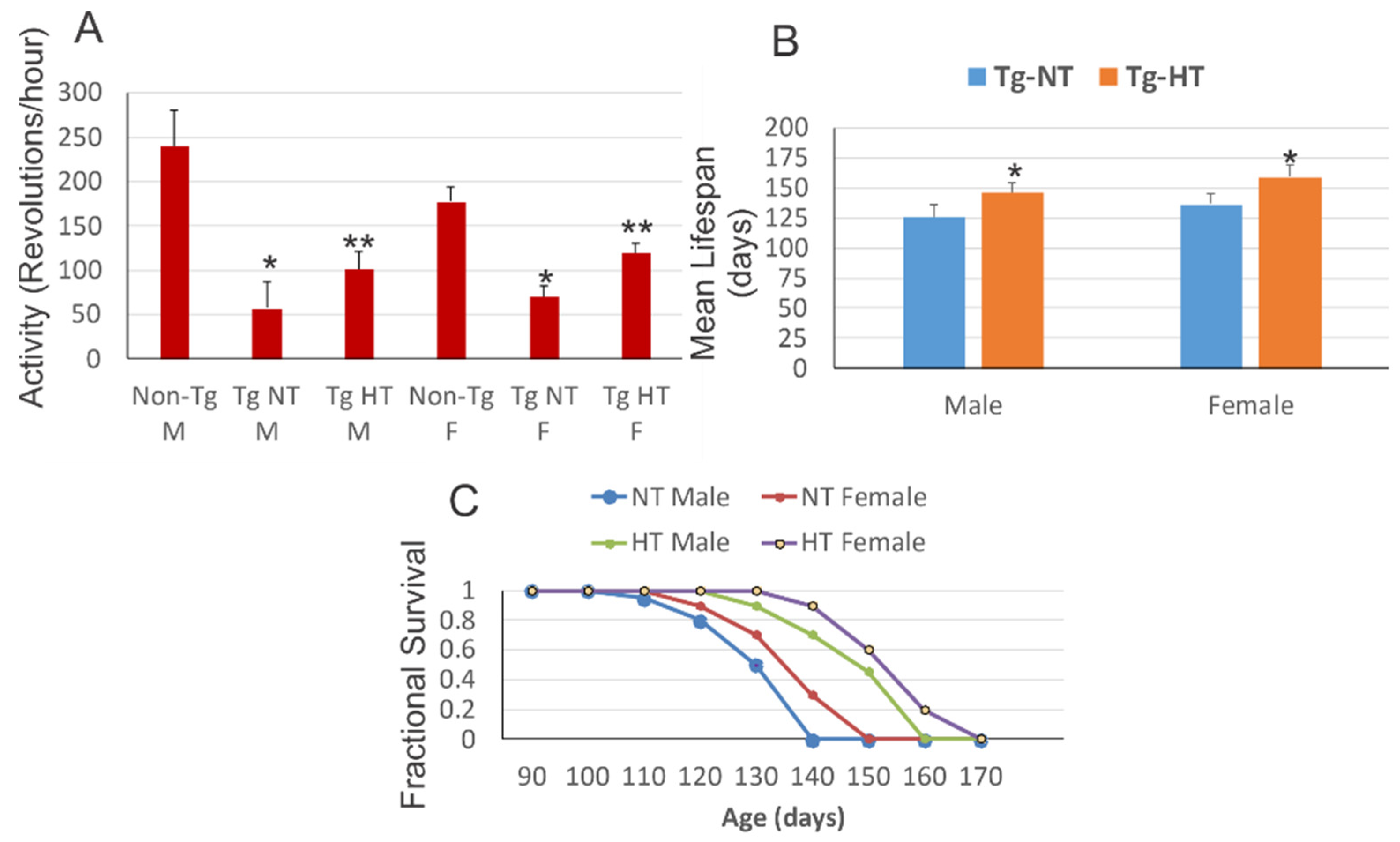
À 10 semaines d'âge, l'hypothermie a amélioré les résultats neurologiques et la survie du modèle de souris SOD1. Les modèles de souris SOD1 mâles et femelles logés à température ambiante présentaient une réduction significative de l'activité motrice par rapport aux compagnons de portée non transgéniques appariés au sexe. Les souris SOD1 mâles acclimatées au froid et les souris SOD1 femelles ont montré une activité motrice significativement améliorée par rapport au modèle de souris SOD1 maintenu à une température normale.
Cependant, tous les groupes de souris SOD1 présentaient des déficits moteurs significatifs par rapport aux souris non transgéniques du même âge. La durée de vie moyenne du modèle de souris SOD1 mâles acclimatées au froid a été augmentée de manière significative par rapport au modèle de souris exposé à une cempérature normale. L'acclimatation au froid du modèle de souris SOD1 femelles a obtenu un résultat légèrement meilleur sur l'augmentation significative de la durée de vie par rapport au modèle de souris SOD1 femelle à température normale. 3.4.
Les auteurs ont évalué les souris SOD1 en hypothermie et à température normale à 12 semaines d'âge pour la neuropathologie de la moelle épinière. Les souris non transgéniques avaient des neurones moteurs spinaux évidents avec de grands corps cellulaires multipolaires. Dans les souris SOD1 non refroidi, les motoneurones ont été épuisés de manière significative à 75% et il y avait une infiltration fulminante secondaire de petites cellules dans le parenchyme. Dans les souris SOD1 acclimatées au froid, les motoneurones étaient plus apparents que les souris non refroidies.
L'hypothermie a sauvé nombre de motoneurones dans la moelle épinière lombaire du modèle de souris SOD1 (perte de 40%) par rapport à la perte de 80% dans les souris SOD1 non refroidi. L'inflammation secondaire à petites cellules du parenchyme de la moelle épinière est apparue atténuée chez les souris acclimatées au froid.
Dans les souris SOD1 non refroidi, les mitochondries étaient sévèrement enflées, dysmorphiques et perturbées dans les motoneurones et dans le neuropile. Dans les souris SOD1 en hypothermie, les mitochondries dans les corps cellulaires des neurones moteurs ont été protégées du gonflement. Cette protection des motoneurones chez les souris SOD1 a été mise en parallèle par une atténuation des changements inflammatoires de la moelle épinière.
Chez les souris non transgéniques, l'innervation de la plaque motrice du diaphragme était proche de 100%, tandis que chez les souris SOD1 non refroidies, l'innervation de la plaque d'extrémité était réduite de manière significative à seulement environ 40% à l'âge de 12 semaines.
En revanche, dans les souris SOD1 acclimatées au froid, l'innervation de la jonction neuromusculaire a été restaurée à environ 65% de la normale, mais était encore significativement réduite par rapport à l'innervation du diaphragme de souris non transgénique.
Les souris SOD1 refroidies présentaient une régulation à la hausse significative des protéines HSP70, UCP3 et SUMO1 par rapport aux souris transgéniques non refroidies.
L'acclimatation au froid était plus efficace chez les femelles que chez les mâles pour prolonger la durée de vie. L'innervation du diaphragme des plateaux moteurs a été améliorée par l'hypothermie.
De nombreuses cytokines circulantes induisent une fièvre. Ce profil de cytokines inflammatoires est cohérent avec leur découverte selon laquelle les souris SLA sont fébriles au cours de l'évolution de la maladie. Les principales cytokines pyrogènes proinflammatoires circulantes chez les souris SLA sont le TNFα et l'IL6. Le TNFα et l'IL6 peuvent entraîner une atrophie musculaire dans divers contextes cliniques. La fonte musculaire squelettique est une caractéristique importante de la SLA humaine et de certains modèles murins de la SLA.
Les auteurs ont confirmé, ce qui avait été rapporté à plusieurs reprises, qu’au cours de l'évolution de la maladie dans les muscles de souris SOD1, la production de monoxyde d'azote est augmentée et la nitration des protéines est élevée, y compris les protéines clés au niveau de la jonction neuromusculaire.
Le monoxyde d'azote est un régulateur essentiel de l'apoptose cellulaire. Il peut avoir un effet antiapoptotique, ou, inversement, un effet apoptotique. Cette bascule est intimement liée à la présence ou non de réducteurs cellulaires tels de glutathion.
Lors de la SLA, la fonction thermorégulatrice pourrait être aberrante. L'hypothermie pourrait agir au niveau du SNC, du système nerveux périphérique, du muscle squelettique et des niveaux de graisse corporelle.
L'application clinique-translationnelle et les tests d'efficacité de ce concept dans la SLA humaine seraient non invasifs et applicables, peut-être dans des spas de réadaptation en milieu privé et hospitalier. L'immersion en eau froide ou la cryothérapie est courante chez les sportifs. Les protocoles d'acclimatation au froid devraient être déterminés et affinés empiriquement, et des biomarqueurs d'efficacité thérapeutique doivent être identifiés pour la SLA humaine. Les biomarqueurs pourraient être basés sur la biopsie de muscles squelettiques et le dosage de HSP70, de la sumoylation des protéines et des seuils d'activation du mPTP.



