Tous les mois une découverte révolutionnaire ?
Tous les mois la presse spécialisée nous informe d'un progrès décisif dans le traitement de la SLA.
Au début du mois de février, c'était un jeune docteur en Écosse, qui s'avère n'avoir fait que des expériences in-vitro montrant un rallongement des axones de neurones moteurs inférieurs quand ils étaient soumis à un inhibiteur de PGC1α, mais le service de presse de son université avait traduit cela comme étant une grande découverte permettant d'espérer l'arrivée prochaine d'un médicament.
On sait depuis 2008 que PPAR-γ est un facteur de transcription qui interagit avec PGC-1α et qu’ils sont impliqués dans la SLA.
Nous sommes à la fin du mois, et voici une nouvelle annonce de découverte importante concernant un traitement de la SLA. Il s'agit d'une molécule qui est développée depuis presque une dizaine d'années à la Northwestern University, dans l'Illinois. Plusieurs articles ont déjà été écrits à propos de cette molécule, mais il s'agissait de trouver une molécule qui soit à la fois caractérisée par une faible toxicité, capable de passer la barrière hémato-encéphalique et d'avoir une action sur la progression de la maladie. Mais en 10 ans il n'avait pas été procédé à des essais extensifs de cette molécule sur des souris modèles de la SLA. Il semblerait que ce soit le cas aujourd'hui, peut-être grâce à la collaboration entre deux équipes, celles du professeur Richard B. Silverman qui est orientée "chimie" et celle de la professeure Hande Ozdinler plutôt orienté « biologie ».
Il y a à la fois une bonne et une mauvaise nouvelle dans ce nouvel article. La mauvaise nouvelle est que la molécule ne serait efficace que sur les neurones moteurs supérieurs. Cela est un peu surprenant et il n’y a pas vraiment d’explications élaborées qui seraient fournis par les auteurs de l’article. En général dans la SLA les deux types de neurones sont affectés et restaurer le fonctionnement des neurones moteurs supérieurs ne sera guère utile, si les neurones moteurs inférieurs sont incapable de commander les muscles. De plus dans l’article il n'est question que de restaurer la bonne santé de neurones malades, donc traités dès l'apparition des symptomes.
Les auteurs ont un langage très agressif
Les auteurs estiment que les autres chercheurs n’ont pas cherché à élaborer des thérapies pour les neurones moteurs supérieurs. C’est évidemment faux, nombres d’études utilisant des échantillons de cerveaux de malades décédés sont là pour en témoigner.
Les auteurs n’hésitent pas non plus à affirmer que les autres chercheurs effectuent leurs recherches in-vitro avec un ensemble différent de lignées cellulaires, parfois sans rapport avec la biologie des motoneurones.
Dans la mesure où le système nerveux implique un grand nombre de types différents de cellules, et que celles-ci sont originaires de seulement quelques lignées de cellules souches, au contraire cela semble très sain. Par ailleurs on pourrait contrer cette focalisation sur les neurones en rappelant que les neurones n’exercent pas leur fonction de façon isolée, un nerf c’est un ensemble de cellules qui collaborent à la même tâche. D'ailleurs à la lecture de l'article on se demande immédiatement pourquoi les auteurs n'ont pas examinés les astrocytes, qui ne sont présent que dans le système nerveux central.
Ils expliquent encore qu’il n'y a jamais eu d'étude qui examine les motoneurones supérieurs au niveau cellulaire au cours de la maladie. Le nombre d’études sur la SLA utilisant des souris modèles de la maladie est là pour témoigner de la fausseté de cette affirmation.
Une thérapie qui est efficace quel que soit le type de proteopathie.
La bonne nouvelle, apparemment inattendue, c’est que la molécule est efficace à la fois pour les pathologies de type SOD1 (environ 2 % des malades) et TDP-43 (environ 95 % des malades).
Les auteurs estiment qu’il s'agit de la première étude axée sur les mécanismes et les cellules et qui jette les bases des études futures qui identifieront les composés en fonction de leur capacité à restaurer la santé des neurones.
Dans cette étude, quatre fines tranches de cortex moteur issus de sujets témoins normaux sans maladie neurologique et neuf tranches issus de patients SLA ont été inclus. Là encore le nombre est très faible, dans d’autres études plusieurs centaines de tissus issus de personnes décédés sont étudiés. Il y a d’ailleurs des banques de données qui permettent de partager les données entre chercheurs.
Coupes histologiques de patients décédés
Les motoneurones supérieurs n'ont été comptés que si leur soma et leur dendrite apicale étaient tous deux visualisés dans la même section de 50 µm d'épaisseur. Cela suppose une sélection sévère des neurones, est-ce que les neurones sélectionnés sont représentatifs de la population de motoneurones supérieurs?.
Identification du NU-9 :
Le cyclohexane chiral 1,3-Diones, est une molécule de cyclohexane contenant deux groupes cétone.
Comme dit plus haut, en fait les travaux sur NU-9 ont débutés il y a presque une dizaine d’années.
Un criblage à haut débit de plus de 50 000 molécules avait été réalisé à l'aide d'un test à base de cellules PC12 exprimant la SOD1G93A pour identifier les composés qui atténuaient l'agrégation des protéines et la cytotoxicité.
Cela a permis d’identifier trois familles de composés :
Un premier composé a été sélectionné parmi plus de 50 analogues de la cyclohexane-1,3-dione, à cause de sa capacité à réduire la toxicité médiée par mSOD1 et à inhiber l'agrégation des protéines.
Plusieurs cycles d'optimisation ont été effectués, aboutissant à un autre composé, qui a également avait d'excellentes propriétés pharmaceutiques in vitro, mais ne pénétrait pas dans les neurones. D'autres modifications de ce composé ont alors conduit à la génération de NU-9, qui pénètre les neurones corticaux, traverse la barrière hémato-encéphalique et possède des propriétés pharmacocinétiques favorables.
Gestion des souris
Les souris sont issues d’un croisement de souris modèle de la SLA de type SOD1 et de souris porteuses d’une protéine fluorescente.
Dans cette étude, seules les souris femelles ont été utilisées pour les expériences.
L’étude porte sur un très petit nombre de souris, on peut se demander si cela à un intérêt de faire une étude sur un nombre de souris aussi faible, car aucun lissage ne permet de gommer les inévitables mesures non significatives. Les souris surréagissent notoirement à d’infimes variations dans leur environnement, il y a peu nous avons ainsi rapporté que des souris modèles de la SLA devenaient malades à Stanford mais pas au MIT !
- Dix souris WT-UeGFP et six hSOD1G93A-UeGFP ont été traitées avec le placebo,
- Cinq souris WT-UeGFP et sept souris hSOD1G93A-UeGFP ont été traitées avec 20 mg / kg / jour dose de NU-9.
- Onze souris WT-UeGFP , neuf souris hSOD1G93A-UeGFP et quatre souris prpTDP-43A315T-UeGFP ont été traitées avec une dose de 100 mg / kg / jour de NU-9.
La dose de NU-9 ou de placebo a été administrée une fois par jour par gavage oral, en commençant au jour postnatal (P) 60 et en continuant jusqu'à P120.
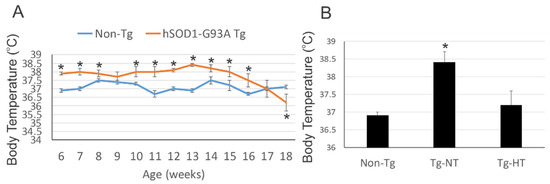
Ce composé NU-9 est capable de prolonger la durée de vie d’une souris modèle de la SLA de 13% avec une dose de 20 mg/kg. Une étude toxicologique répétée pendant 7 jours chez la souris a mis en évidence une dose sans effet nocif observé de 100 mg/kg.
Toutes les souris ont été sacrifiées au bout de 120 jours (P120), ce qui est considéré comme un stade terminal pour ce type de souris modèle de la SLA et environ 60% des motoneurones supérieurs dans le cortex moteur sont alors perdus tandis que les motoneurones supérieurs restants ont une taille de soma plus petite et des dendrites apicales vacuolées et désintégrées. Il ne semble pas y avoir eu de test sur un accroissement de durée de vie des souris traitées.
Analyses qualitatives SOD1
Le microscope électronique a permis des analyses du type de cellule des motoneurones supérieurs et de leurs organites clés avec une grande précision. À P120, les motoneurones supérieurs des souris hSODG93A-UeGFP traitées avec le placebo avait perdu la plupart de leur intégrité cytoplasmique. Il y restait très peu d'organites intacts dans le soma. Cependant, la présence de mitochondries et de réticulum endoplasmique désintégrés étaient évidentes. Les mitochondries avaient principalement perdu l'intégrité de leur membrane interne, s’étaient agrégées, agrandies ou avaient commencé à se désintégrer. Le réticulum endoplasmique montrait également des citernes brisées et dispersées.
Le traitement NU-9 (dose de 100 mg / kg par jour) a montré de profondes améliorations à la fois de la structure et de l'intégrité des mitochondries et du réticulum endoplasmique des motoneurones supérieurs malades. Lors du traitement, le soma de ces neurones montrait une membrane nucléaire intacte, qui était dépourvue de toute invagination ou saillie, et la détection de nombreux organites qui étaient appropriés en taille, emplacement et interactions entre eux. La membrane interne mitochondriale était intacte avec des crêtes appropriées qui étaient en contact étroit avec le réticulum endoplasmique.
Analyses quantitatives SOD1
Les auteurs ont ensuite effectué des analyses quantitatives pour déterminer si ces améliorations étaient largement observées dans les motoneurones supérieurs malades traités avec NU-9. Le nombre total de mitochondries dans les motoneurones supérieurs de souris hSODG93A-UeGFP avait augmenté de manière significative après un traitement de 100 mg / kg / jour NU-9 par rapport aux motoneurones supérieurs traités avec le placebo. Le nombre total de mitochondries après traitement NU-9 était comparable à celui des souris saines. De plus, le traitement NU-9 a augmenté de manière significative le nombre de mitochondries et de réticulum endoplasmique sains dans les motoneurones supérieurs.
Et surtout, le nombre moyen d'motoneurones supérieurs présents dans le cortex moteur des souris hSOD1G93A-UeGFP traitées 60 jours avec 100 mg / kg / jour de NU-9 est devenu presque comparable à celui des nombres d'motoneurones supérieurs présents chez les souris saines
Et TDP-43 ?
Étant donné que le NU-9 semble améliorer l'intégrité des mitochondries et du réticulum endoplasmique des motoneurones supérieurs malades à cause de la toxicité de mSOD1, et qu'il s'agit de problèmes que l’on trouve aussi dans les pathologies de type TDP-43, les scientifiques ont supputé que NU-9 pourrait aussi être efficace dans la pathologie TDP-43. Cela semble quand même un grand pas conceptuel, en effet il s'agit de deux modèles de maladie distincts. Même si NU-9 a un effet sur la structure des mitochondries et du réticulum endoplasmique des motoneurones supérieurs, il ne devrait pas agir sur les agrégats de protéines mal formées et mal localisées ?
Pour tester leur nouvelle hypothèse, les scientifiques ont généré un nouveau type de souris, cette fois-ci modélisant la SLA de type TDP-43.
Quatre souris ont été traitées avec une dose de 100 mg / kg / jour de NU-9, et 3 souris de type prpTDP-43A315T-UeGFP ont été utilisées comme contrôle. La cohorte de souris WT-UeGFP a été utilisée comme témoin sain pour les deux groupes.
Analyses qualitatives TDP-43
Le traitement NU-9 a entraîné de profondes améliorations à la fois des mitochondries et des réticulums endoplasmique des motoneurones supérieurs. Les mitochondries, en particulier leurs membranes internes, sont devenues intactes et il n'y avait aucun signe de mitoautophagie ou de mitophagie. Le réticulum endoplasmique conservait sa structure avec des ribosomes attachés, et il n'y avait pas d'élargissement ou de désintégration des citernes.
Analyses quantitatives TDP-43
L'analyse quantitative a confirmé une augmentation significative du nombre de mitochondries totales par motoneurones supérieurs par section après 100 mg / kg / jour de traitement NU-9. Ce nombre de mitochondries après traitement NU-9 chez les souris prpTDP-43A315T-UeGFP est devenu comparable à celui des souris saines. Le pourcentage moyen de mitochondries saines a également augmenté de manière significative avec le traitement NU-9 par rapport aux motoneurones supérieurs malades. Le traitement NU-9 a également augmenté le nombre moyen de cisternes de réticulum endoplasmique intactes dans les motoneurones supérieurs.
Les scientifiques ont ensuite examiné si NU-9 le traitement favoriserait également l'intégrité cellulaire et la survie de l'motoneurones supérieurs avec la pathologie TDP-43 in vivo. La santé et l'intégrité des dendrites apicales chez les souris prpTDP-43A315T-UeGFP ont montré une amélioration profonde avec le traitement NU-9 car le pourcentage d'motoneurones supérieurs avec des dendrites apicales primaires vacuolées était significativement réduit. Plus intéressant encore, le nombre moyen d'motoneurones supérieurs dans le cortex moteur des souris prpTDP-43A315T-UeGFP traitées avec NU-9 a connu une augmentation profonde par rapport à celui des souris prpTDP-43A315T-UeGFP non traitées.
La même cohorte WT-UeGFP a été utilisée comme témoin sain pour les souris hSOD1G93A-UeGFP et prpTDP-43A315T-UeGFP, comme mentionné précédemment. Les nombres motoneurones supérieurs avec le traitement NU-9 étaient comparables et presque identiques à ceux des souris témoins saines traitées avec le placebo, révélant la capacité du NU-9 à éliminer la dégénérescence continue des motoneurones supérieurs.
Effet du NU-9 sur les neurones moteurs inférieurs
Dans le but de déterminer si le traitement NU-9 améliore également la santé et la survie des neurones moteurs inférieurs (motoneurones inférieurs), les auteurs ont étudié les cordons lombaires des souris hSOD1G93A-UeGFP et prpTDP-43A315T-UeGFP. Comme indiqué par les études précédentes, il n'y avait pas de perte importante de motoneurones inférieurs dans la moelle épinière des souris prpTDP-43A315T-UeGFP, même à P120, et donc une enquête sur le traitement NU-9 sur la survie des motoneurones inférieurs n'était pas possible. Cela semble indiquer que la pathologie TDP-43 n’affecterait que les motoneurones supérieurs, pourtant d’autres études montrent le contraire.
Cependant, dans le cas de SOD1, il y a eu une réduction spectaculaire du nombre de motoneurones inférieurs chez les souris hSOD1G93A-UeGFP par rapport aux souris saines.
Effets macroscopiques
Les auteurs ont évalué quantitativement les changements dans le nombre de motoneurones inférieurs dans la moelle épinière lombaire des souris qui ont été traitées soit avec le placebo, soit NU-9 (20 ou 100 mg / kg / jour), ou encore les souris saines témoins. Le traitement NU-9, quelle que soit la dose, n'était pas suffisant pour éliminer la dégénérescence motoneurones inférieurs en cours chez les souris hSOD1G93A-UeGFP.
Le traitement NU-9 améliore la fonction des neurones moteurs supérieurs
Même si la plupart des tests comportementaux ne parviennent pas à évaluer correctement la santé et la connectivité motoneurones supérieurs, le test du fil suspendu serait plus spécifique à l'intégration motoneurones supérieurs.
Les souris hSOD1G93A-UeGFP non traitées n'ont pas pu rester sur le fil de suspension au fur et à mesure que la maladie progressait. Au contraire, les souris hSOD1G93A-UeGFP traitées avec une dose de 100 mg / kg / jour de NU-9 ont obtenu des résultats meilleurs que les souris hSOD1G93A-UeGFP traitées avec le placebo, et cette performance était comparable à celle de souris saines au même âge. Cependant le traitement au NU-9 n'a pas entraîné d'amélioration significative des performances au test du rotarod, quelle que soit la dose.
Les souris prpTDP-43A315T ont obtenu de moins bons résultats que les compagnons de portée WT aux tests des rotarod et des fils suspendus. Cependant, lorsqu'elles ont été traitées avec une dose de 100 mg / kg / jour de NU-9, elles ont obtenu de meilleurs résultats au test du fil suspendu, comparables à ceux des souris WT saines à P120. A la différence du modèle de souris hSOD1G93A, même le test Rotarod a révélé une amélioration significative du modèle TDP-43 seulement après 30 jours de traitement NU-9.
Conclusion
La capacité du NU-9 à améliorer l'intégrité à la fois des mitochondries et du réticulum endoplasmique est significative, car, même si les causes sous-jacentes de la maladie sont hétérogènes, de nombreuses indices convergent vers le bon fonctionnement des mitochondries et du réticulum endoplasmique. La perturbation des organites membranaires intracellulaires, telles que l'appareil de Golgi, a été suggérée comme une cause possible de l'ALS et est proposée comme étant en amont du dysfonctionnement du réticulum endoplasmique.
Cela pourrait expliquer pourquoi le traitement NU-9 améliore la cytoarchitecture motoneurones supérieurs et élimine leur dégénérescence progressive chez les souris hSOD1G93A et TDP-43A315T.
Cependant cela n’explique pas pourquoi les neurones moteurs inférieurs ne seraient pas affectés par cette molécule. L’explication donnée, qu’il s’agit de lignées différentes, semble très courte et n’a pas fait l’objet de plus d’investigation. A aucun moment on ne parle d’axones dans cet article, ce qui semble bizarre dans un article qui annonce chercher un remède à la SLA. Il me semble que cela remet en cause l’affirmation que le traitement NU-9 améliore la cytoarchitecture motoneurones supérieurs.
Par ailleurs le nombre de souris testées est très, très faible et probablement sans signification sur le plan statistique. Il en est de même pour les quelques coupes histologiques de patients décédés, et de la sélection de neurones moteurs. On peut aussi remarquer qu'il est démontré depuis plus de 15 ans que d'autres types de cellules sont impliqués dans la SLA, à commencer par les astrocytes, que les motoneurones. Cela n'a absolument pas été étudié dans cet article. Pourtant qu'une molécule soit efficace sur les motoneurones supérieurs et pas sur les motoneurones inférieurs fait immédiatement penser que ce n'est pas sur les neurones qu'elle agit mais sur les astrocytes.
Tout cela relativise considérablement les affirmations très fortes du service de presse de l’université qui rapportait que « After 60 days of treatment, diseased brain cells look like healthy cells » et les négations de la valeur du travail des autres scientifiques qui parsème l’article.
Pour toute correspondance ecrivez moi à contact [at] padiracinnovation.org
Publicité

Ce livre en Anglais retrace les principales réalisations de la recherche sur la SLA au cours des 30 dernières années. Il présente les médicaments en cours d’essai clinique ainsi que les recherches en cours sur les futurs traitements susceptibles d’ici quelques années, d’arrêter la maladie et de fournir un traitement complet en une décennie ou deux.
 L'AVC est une des principales causes de décès et la principale cause d'invalidité de longue durée. Les patients survivant à un AVC présentent un risque majeur de développer plus tard une démence vasculaire et/ou de type Alzheimer.
Ce risque est particulièrement élevé chez les personnes âgées car divers processus cellulaires sont altérés au cours du vieillissement.
L'AVC est une des principales causes de décès et la principale cause d'invalidité de longue durée. Les patients survivant à un AVC présentent un risque majeur de développer plus tard une démence vasculaire et/ou de type Alzheimer.
Ce risque est particulièrement élevé chez les personnes âgées car divers processus cellulaires sont altérés au cours du vieillissement.