Une avancée intéressante en neurosciences régénératives
Le texte présenté dans ce post décrit une avancée majeure en neurosciences régénératives : la possibilité de transformer un type spécifique de cellules de soutien cérébrales en neurones corticospinaux (NCS) hautement spécifiques et fonctionnels.
Ces neurones, également appelés motoneurones supérieurs, sont ceux qui meurent dans la SLA (maladie de Charcot) ou qui sont sectionnés lors de lésions de la moelle épinière, entraînant une paralysie permanente.
On sait depuis plus d'une dizaine d'années faire maturer in vitro des cellules souches vers des cellules matures, c'est-à-dire différenciées en morphologie et en rôle. On sait même transformer des cellules matures en cellules souches. Mais il s'agit là de « preuves de concept » ; le chemin est encore long pour transformer cela en thérapies commercialisables à grande échelle.
Le concept reste toutefois fascinant : au lieu de micro-actions chimiques sur des cellules vieillissantes, il s'agit de favoriser la création de cellules de remplacement jeunes. De plus, la SLA atteint des cellules qui ne sont jamais remplacées au cours de la vie : les neurones moteurs et les fibres des muscles squelettiques.
1. Résumé des principaux résultats
La recherche a identifié une population cellulaire spécifique, organisée en mosaïque dans le cerveau, appelée progéniteurs SOX6+/NG2+. Alors que ces cellules agissent généralement comme précurseurs des cellules gliales (cellules de soutien), l’étude révèle qu’elles sont « préparées au développement » — en quelque sorte préprogrammées — à devenir des neurones si elles reçoivent l’impulsion moléculaire adéquate.
Le cocktail « NVOF »
Les chercheurs ont développé un outil génétique multicomposant appelé NVOF (Neurog2, VP16-Olig2 et Fezf2). La population cellulaire spécifique identifiée dans cet article mature normalement en cellules gliales, c'est-à-dire des cellules de taille habituelle (quelques microns de diamètre), alors qu'un neurone moteur possède un axone pouvant atteindre plusieurs centaines de fois cette longueur. De plus, le rôle des cellules gliales n'est pas celui de neurone, mais de soutien et d'assainissement de la population neuronale.
Contrairement aux méthodes précédentes qui créaient des neurones « génériques », ce cocktail spécifique active le programme neurogénique dormant et supprime la voie naturelle de différenciation gliale. Le cocktail oriente les cellules vers un phénotype similaire à celui des neurones corticospinaux (NCS), avec des axones longs et des propriétés électriques caractéristiques.
2. Contexte : L’importance de la spécificité
Le texte souligne que remplacer des neurones par des versions génériques ne suffit pas. Dans les maladies neurodégénératives, seuls des sous-types neuronaux très spécifiques meurent (vulnérabilité sélective). Or, de nombreux neurones cultivés en laboratoire sont « confus » : ils expriment simultanément des marqueurs de plusieurs types cellulaires différents, ce qui les rend inadaptés comme médicaments contre la SLA.
L'avantage d'utiliser des progéniteurs locaux réside dans l'exploitation des cellules SOX6+/NG2+ déjà présentes dans le cortex, plutôt que de devoir transplanter des cellules souches, car :
Elles partagent le même site d’origine et la même lignée que les neurones perdus.
Elles n’ont pas besoin de migrer sur de longues distances dans le cerveau.
Elles sont déjà intégrées dans tout le cortex, prêtes à être reprogrammées précisément là où elles sont nécessaires.
De plus, la transplantation de cellules pose de nombreux problèmes et s'avère en général d'une très faible efficacité. Cette nouvelle publication ne se contente pas de décrire les gènes utilisés, elle se concentre sur la cinétique (la vitesse et la durée du signal) et détaille quand et comment les instructions génétiques sont transmises aux cellules. L'idée principale est que, pour créer un neurone sain, on ne peut pas simplement bloquer l'activation ; il faut imiter le rythme naturel du développement.
3. La méthode de transmission « hybride »
Les chercheurs ont constaté que différents gènes nécessitent différents mécanismes d'activation. Certains requièrent une activation rapide, tandis que d'autres nécessitent un flux continu. Le problème est que maintenir le gène Neurog2 (l'activateur) actif trop longtemps est toxique pour la cellule.
Pour résoudre ce problème les chercheurs ont utilisé de l'ARNm modifié. Contrairement à l'ADN, qui peut persister ou s'intégrer au génome, l'ARNm est transitoire. Il transmet les instructions, la cellule synthétise les protéines, puis l'ARNm se dégrade naturellement. C'est un mécanisme proche de celui des vaccins contre la Covid-19. Il se produit un pic d'expression protéique important en 12 à 24 heures, qui disparaît ensuite, imitant le comportement du gène chez l'embryon.
Le « flux continu » : ADN plasmidique (Fezf2)
Le gène Fezf2 (le promoteur de l'identité neuronale) doit rester actif tout au long de la vie du neurone. La solution a été d'utiliser de l'ADN plasmidique sous le contrôle d'un promoteur constitutif (appelé CAG), maintenant le gène actif indéfiniment.
Résultat : La cellule reçoit une impulsion temporaire pour devenir un neurone (via l'ARNm), mais aussi une instruction permanente pour rester un motoneurone (via l'ADN).
4. Sécurité et validation
Un point crucial est que la méthode par ARNm évite l'intégration dans l'ADN de la cellule cible. En thérapie génique traditionnelle, il existe un risque d'insertion d'ADN à un endroit inapproprié du génome (pouvant provoquer un cancer). L'usage de l'ARNm élimine ce risque d'altération génomique permanente, un indice majeur pour la sécurité de futurs essais cliniques.
Pour démontrer la faisabilité du traitement simultané, les chercheurs ont utilisé des marqueurs colorés :
La GFP (protéine fluorescente verte) pour l'impulsion (ARNm).
La tdTomato (protéine rouge) pour le flux continu (ADN). L'observation de cellules devenant à la fois rouges et vertes a confirmé qu'un seul traitement permet de délivrer des instructions complexes et synchronisées.
5. Thérapies futures : pistes et implications
Ce texte évoque une transition de la simple gestion des maladies cérébrales vers une réparation in situ (sur place). Cette avancée ouvre la voie à :
La création de cultures « pures » de NCS pour identifier des médicaments capables de stopper spécifiquement la dégénérescence.
L'utilisation d'ARNm synthétique pour stimuler les cellules NG2 du patient afin de les transformer en nouveaux motoneurones, sans chirurgie lourde ni transplantation.
Les auteurs soulignent que, bien que ces nouveaux neurones soient très proches des naturels, ils ne sont pas encore parfaits (présentant parfois des traits de neurones cortico-thalamiques). Les thérapies futures impliqueront probablement l'ajout de facteurs de croissance ou un ajustement plus fin du timing génétique pour garantir une maturité totale.
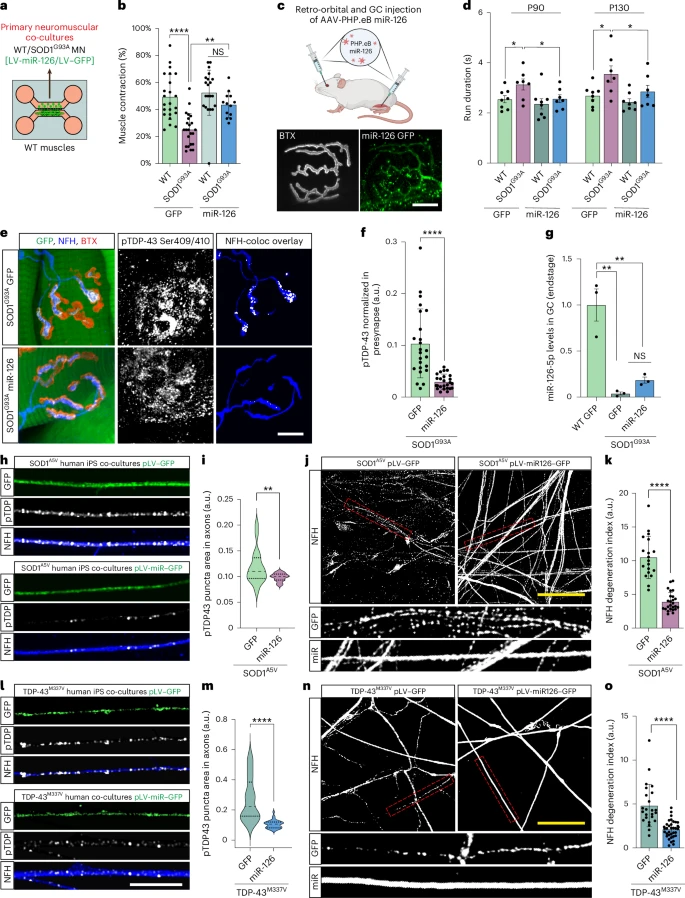 When pathologists examine the spinal motor neurons of patients with SOD1-related ALS, the nuclei generally appear normal: the TDP-43 protein is always present, and abnormal aggregates are rarely observed. This is why SOD1-related ALS has been considered "TDP-43 negative."
When pathologists examine the spinal motor neurons of patients with SOD1-related ALS, the nuclei generally appear normal: the TDP-43 protein is always present, and abnormal aggregates are rarely observed. This is why SOD1-related ALS has been considered "TDP-43 negative." Analyses statistiques contestables
Analyses statistiques contestables

