A new publication by Jamie K Wong, and colleagues including two well known ALS scientists argues apolipoprotein B-100 in sporadic amyotrophic lateral sclerosis CSF is the putative agent responsible for inducing motor disability, motor neuron degeneration and pathological translocation of TDP-43.
While there are publications with similar claims about every week, this one sounds impressive, the scientists here have really worked hard to make sure they haven't left any stone unturned.
For a layperson like me, at first glance, this publication makes sense as there is a special relation between ALS and lipid metabolism. Apolipoproteins are proteins that bind lipids (oil-soluble substances such as fats, cholesterol and fat soluble vitamins) to form lipoproteins. They transport lipids in blood, cerebrospinal fluid and lymph. There are multiple classes of apolipoproteins and several sub-classes
ApoD level increases in nervous system with a large number of neurologic disorders inclusive of Alzheimer's disease, schizophrenia, and stroke. ApoE has been implicated in dementia and Alzheimer's disease.
So why not ApoB and ALS? Moreover overproduction of apolipoprotein B can result in lipid-induced endoplasmic reticulum stress and insulin resistance in the liver. ER stress leads to mislocalized misfolded proteins in cytosol, and half of ALS patients exhibit insulin resistance. In addition patients with ALS have higher levels of LDL-C, ApoB, and ApoB/ApoAI ratio already 20 years before diagnosis.
Yet this is not a study on humans but on mice and motor neurons in-vitro, and as usual a lot could be said about mice animal models of ALS. So maybe their claim, that ApoB is the agent responsible for inducing sporadic ALS, needs more work.
In addition a recent publication claimed that ALS patients that have elevated levels of ApoB in blood are associated with a lower risk of death. ApoB is also correlated with LDL (the "bad" cholesterol). Another study claimed the contrary. Yet another study did not find any evidence of association between lipoprotein or apolipoprotein levels and clinical findings.
There is also a lack of associations of cholesterol-lowering drugs (which lowers ApoB), antihypertensive drugs, and antidiabetics with the risk of ALS.
So we must remain cautious again, anyway even if this finding about ApoB and ALS was true, a commercial drug would be available only in 10 or 20 years.
 Three important discoveries showed how these mental maps were implemented in the mammalian brain.
* The first, in the early 1970s, is that hippocampal neurons, called place cells, respond to the position of the animal.
* The second, in the early 1990s, is that neurons in neighboring regions, called head direction cells, respond in the direction of the animal's head. This makes it possible to manipulate movement information and see how the location and lead direction cells react.
* The third finding was that the organization of neurons in the dorsomedial entorhinal cortex, named grid cells, closely resemble a sheet of squared paper organized in a hexagonal fashion and suggests that place cells can use grid cells to calculate distances.
Three important discoveries showed how these mental maps were implemented in the mammalian brain.
* The first, in the early 1970s, is that hippocampal neurons, called place cells, respond to the position of the animal.
* The second, in the early 1990s, is that neurons in neighboring regions, called head direction cells, respond in the direction of the animal's head. This makes it possible to manipulate movement information and see how the location and lead direction cells react.
* The third finding was that the organization of neurons in the dorsomedial entorhinal cortex, named grid cells, closely resemble a sheet of squared paper organized in a hexagonal fashion and suggests that place cells can use grid cells to calculate distances.
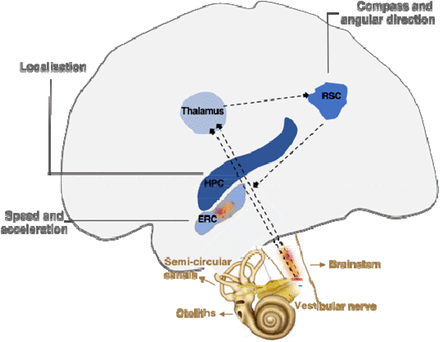 Deficits of path integration, ie of these mental maps, manifest themselves at the onset of Alzheimer's disease. Decades before the expected onset of the disease subtle changes in pathway integration are also present in adults at genetic risk for Alzheimer's disease.
Deficits of path integration, ie of these mental maps, manifest themselves at the onset of Alzheimer's disease. Decades before the expected onset of the disease subtle changes in pathway integration are also present in adults at genetic risk for Alzheimer's disease.

 In this article, the authors outline the principles of drug selection for Parkinson disease prevention trials, focused on proof-of-concept opportunities that will help establish a methodological foundation for this fledgling field.
In this article, the authors outline the principles of drug selection for Parkinson disease prevention trials, focused on proof-of-concept opportunities that will help establish a methodological foundation for this fledgling field.