Studies in arthropods have revealed the existence of mental maps of their position that are very effective in achieving their objective. These maps make it possible to determine their position and the direction to follow. Scientists call these maps "path integration".
 Three important discoveries showed how these mental maps were implemented in the mammalian brain.
* The first, in the early 1970s, is that hippocampal neurons, called place cells, respond to the position of the animal.
* The second, in the early 1990s, is that neurons in neighboring regions, called head direction cells, respond in the direction of the animal's head. This makes it possible to manipulate movement information and see how the location and lead direction cells react.
* The third finding was that the organization of neurons in the dorsomedial entorhinal cortex, named grid cells, closely resemble a sheet of squared paper organized in a hexagonal fashion and suggests that place cells can use grid cells to calculate distances.
Three important discoveries showed how these mental maps were implemented in the mammalian brain.
* The first, in the early 1970s, is that hippocampal neurons, called place cells, respond to the position of the animal.
* The second, in the early 1990s, is that neurons in neighboring regions, called head direction cells, respond in the direction of the animal's head. This makes it possible to manipulate movement information and see how the location and lead direction cells react.
* The third finding was that the organization of neurons in the dorsomedial entorhinal cortex, named grid cells, closely resemble a sheet of squared paper organized in a hexagonal fashion and suggests that place cells can use grid cells to calculate distances.
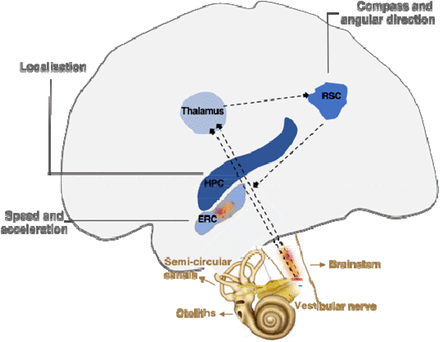 Deficits of path integration, ie of these mental maps, manifest themselves at the onset of Alzheimer's disease. Decades before the expected onset of the disease subtle changes in pathway integration are also present in adults at genetic risk for Alzheimer's disease.
Deficits of path integration, ie of these mental maps, manifest themselves at the onset of Alzheimer's disease. Decades before the expected onset of the disease subtle changes in pathway integration are also present in adults at genetic risk for Alzheimer's disease.
Previous studies of path integration have focused on tasks based on visual cues. The vestibular system is a barosensitive sensory organ, located in the inner ear, which contributes to the sensation of movement and balance in most mammals. So the study of these "path integration" maps must absolutely involve the vestibular system. This is realized in a new study published on MedRxiv, by Gillian Coughlan, Michael Hornberger and their colleagues.
One hundred and fifty participants aged 50-75 were recruited to take part in a research study at the University of East Anglia, Norwich, UK.
Saliva kits were sent to participants home and returned to the university the same day the saliva sample was taken to determine APOE genotype status. Sensor data was collected on the iPad-based assessment tool. The final sample size of 53 included 32 ε3ε3 carriers and 21 cross-sectional ε3ε4 carriers, each of whom completed the background cognitive test and vestibular task on the same day, as well as 3 homozygous APOE-ε4ε4 carriers.
The participants were asked to raise their legs (i.e. without touching the ground) and were turned over by the manipulator. Three seconds after the end of the flip, participants had to point the iPad as precisely as possible in the direction of the starting point, while still wearing the headband and earplugs. The iPad recorded vestibular data: acceleration, rotation and direction.
The scientists' results show impaired vestibular function, a deficiency in people at genetic risk for Alzheimer's disease. Vestibular function differentiated ε3ε4 carriers from ε3ε3 carriers, regardless of demographic background. Machine learning algorithms achieved significant performance in classifying genetic groups based on vestibular function, while univariate statistics failed to identify vestibular differences between APOE groups.
Animal and human studies also suggest a strong anatomical and functional interdependence between the vestibular system and the navigational system. Dysregulation of the vestibular system is associated with deficits in pathway integration.
Vestibular signals that influence pathway integration in preclinical Alzheimer's disease can help identify pathological changes before disease onset and thus guide treatment.
Identifying vestibular contributions to the cognitive phenotype of preclinical Alzheimer's disease is important because vestibular dysfunction is often present with treatable hearing loss. Additionally, vestibular balance training improved spatial orientation in monkeys with severe vestibular damage, suggesting that human adults with vestibular dysfunction, might respond to vestibular implant and/or intensive vestibular training.
Moreover, as the vestibular system has extensive connections with brain regions vulnerable to Alzheimer's disease, including the hippocampus, cingulate cortex, and parietal lobe, vestibular stimulation may indeed improve cognitive performance related to integrity of these brain regions, including disorientation and memory loss due to Alzheimer's disease.


 In this article, the authors outline the principles of drug selection for Parkinson disease prevention trials, focused on proof-of-concept opportunities that will help establish a methodological foundation for this fledgling field.
In this article, the authors outline the principles of drug selection for Parkinson disease prevention trials, focused on proof-of-concept opportunities that will help establish a methodological foundation for this fledgling field.