A Promising Step Forward in Parkinson’s Disease Cell Therapy
There are few new approaches to alleviate Parkinson's disease symptoms. Most of them either consist of supplementing the brain with dopamine or introducing perturbations deep in the brain. As with other neurodegenerative diseases, a regenerative approach is potentially much more attractive. A recent report from the First Affiliated Hospital of the University of Science and Technology of China (USTC) has drawn considerable attention in the field of Parkinson’s disease (PD) research. Led by neurosurgeon Shi Jiong, the team has developed an iPSC-based cell therapy that may mark a meaningful step toward regenerative treatment for PD.
A Highly Efficient Stem Cell–Based Approach
The therapy relies on induced pluripotent stem cells (iPSCs) that are reprogrammed from adult donor cells. These iPSCs are then differentiated into dopamine-producing neural progenitors, the type of cells that gradually disappear in Parkinson’s disease. One of the notable achievements of the USTC team is its >80% efficiency in generating these dopaminergic neurons — a rate well above the standard reported by other groups.
The resulting product, called NCR201 and developed by Nuwacell, is now being evaluated in a Phase I clinical trial. The early results are striking. One participant, who initially had severe motor disability (UPDRS score 62), improved to near-normal function (score 28) within three months of the implant procedure. The researchers describe this as a “functional cure,” while emphasizing that longer follow-up is essential before drawing final conclusions.
A Precise Surgical Method Built for Cell Survival
Delivering fragile stem-cell–derived progenitors into deep brain structures requires meticulous technique. The USTC team uses a combination of three elements designed to maximize precision while minimizing risk:
- Stereotactic Cell Implantation Stereotaxy provides a three-dimensional coordinate system that allows surgeons to target very small structures with sub-millimeter accuracy. For PD, the target is the putamen, part of the striatum, where dopamine normally acts to regulate movement.
- MRI Guidance Instead of relying solely on pre-operative imaging, the team performs the procedure with intra-operative MRI. This allows real-time visualization of the putamen, the cannula tip, and even the distribution of the infused cell suspension. Because brain tissue can shift during surgery, real-time imaging helps ensure that the cells are released exactly where intended.
- Bilateral, Low-Dose Implantation Parkinson’s disease affects both sides of the brain, so the procedure involves implantation into both putamina. The “low-dose” designation reflects the specific cell dose tested in this early trial arm and the minimally invasive nature of the robot-assisted approach.
Together, these components aim to increase both safety (reducing the risk of hemorrhage or misplacement) and efficacy (ensuring cells reach the optimal anatomical location). The dramatic clinical outcome reported in the case example likely reflects this high degree of precision.
What NCR201 Actually Is: Dopaminergic Progenitor Cells from iPSCs
The therapy uses iPSC-derived dopaminergic progenitor cells, a format designed to balance safety and function: • iPSCs: Adult donor cells are reprogrammed into pluripotent cells that can be expanded indefinitely. Using a well-characterized, allogeneic donor line allows for consistency and avoids ethical issues tied to fetal tissue. • Dopaminergic progenitors: Instead of transplanting fully mature neurons, clinicians implant precursor cells that are already committed to the dopamine-producing lineage. This reduces the chance of tumor formation and allows the cells to complete their maturation inside the patient’s brain, where they can better integrate into existing circuits.
The therapeutic goal is straightforward: provide a renewable source of dopamine in the striatum, ideally restoring motor function in a sustained, biological way.
Known Challenges in Early-Stage Stem Cell Trials
As promising as the first results are, Phase I trials are designed to evaluate safety, and several key risks remain central to ongoing monitoring:
Tumor formation from any remaining undifferentiated iPSCs.
Immune rejection, since NCR201 is not patient-specific and may require immunosuppressive medication.
Long-term survival and stable function of the transplanted cells, which must endure for years in the diseased brain environment.
Manufacturing consistency, an ongoing challenge for living cell products.
Other groups worldwide are pursuing similar strategies. Their approach uses donor cells selected for immunological compatibility and a specialized cell-sorting method to enhance safety. Autologous trials, in which each patient receives cells derived from their own tissue, are also under development, though they are slower and more expensive to produce.
Could This Become a Commercial Therapy?
The commercial potential is substantial, especially if long-term data confirm that a single transplant can restore durable motor function. Such an outcome would represent a major advance compared with current PD medications, which provide symptomatic relief but do not modify the course of the disease.
That said, the path to approval is lengthy: • 10–15 years is a realistic estimate before this type of therapy could reach the market. • Phase II and III trials will need to show benefit in larger and more diverse patient populations, with long-term follow-up to rule out late complications. • Manufacturing and surgical delivery will remain complex and costly, at least initially. Scaling these procedures in a way that ensures accessibility will require continued innovation.
Outlook
The early success of the USTC team’s cell-replacement trial is an encouraging sign for regenerative approaches to Parkinson’s disease. It combines a high-quality iPSC-derived cell product with a precise, imaging-guided surgical technique. While many questions remain — especially concerning longevity, safety, and scalability — the work adds meaningful momentum to an area of research that could, over time, change the way PD is treated.
 The authors found that key immune signaling proteins, specifically those containing Death Fold Domains (DFDs) (like ASC, FADD, BCL10, MAVS, TRADD), exist in a unique physical state inside our cells called metastable supersaturation. These full-length adaptors retain nucleation barriers and are able to exist supersaturated in cells. In contrast, many receptors and effectors do not. This localizes the “spring-loaded” behaviour to central adaptors that link receptor sensing to downstream cell-fate decisions.
The authors found that key immune signaling proteins, specifically those containing Death Fold Domains (DFDs) (like ASC, FADD, BCL10, MAVS, TRADD), exist in a unique physical state inside our cells called metastable supersaturation. These full-length adaptors retain nucleation barriers and are able to exist supersaturated in cells. In contrast, many receptors and effectors do not. This localizes the “spring-loaded” behaviour to central adaptors that link receptor sensing to downstream cell-fate decisions. What did the researchers do?
Nine individuals with Parkinson’s, all of whom had deep brain stimulation (DBS) implants, participated in up to 12 cycling sessions over a month. These implants allowed researchers to directly record brain activity from a small structure deep in the brain called the subthalamic nucleus (STN)—a region strongly involved in movement control and a common target in Parkinson’s treatments.
What did the researchers do?
Nine individuals with Parkinson’s, all of whom had deep brain stimulation (DBS) implants, participated in up to 12 cycling sessions over a month. These implants allowed researchers to directly record brain activity from a small structure deep in the brain called the subthalamic nucleus (STN)—a region strongly involved in movement control and a common target in Parkinson’s treatments. The authors note that, although their findings are exploratory and cannot be directly applied clinically yet, the identified drugs could be considered for future clinical trials. Indeed, funding should be sought for these future trials, and since it is almost always private investors who finance clinical trials, they would require more information before making any commitments.
The authors note that, although their findings are exploratory and cannot be directly applied clinically yet, the identified drugs could be considered for future clinical trials. Indeed, funding should be sought for these future trials, and since it is almost always private investors who finance clinical trials, they would require more information before making any commitments.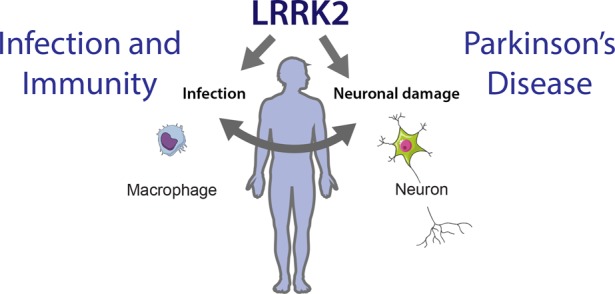
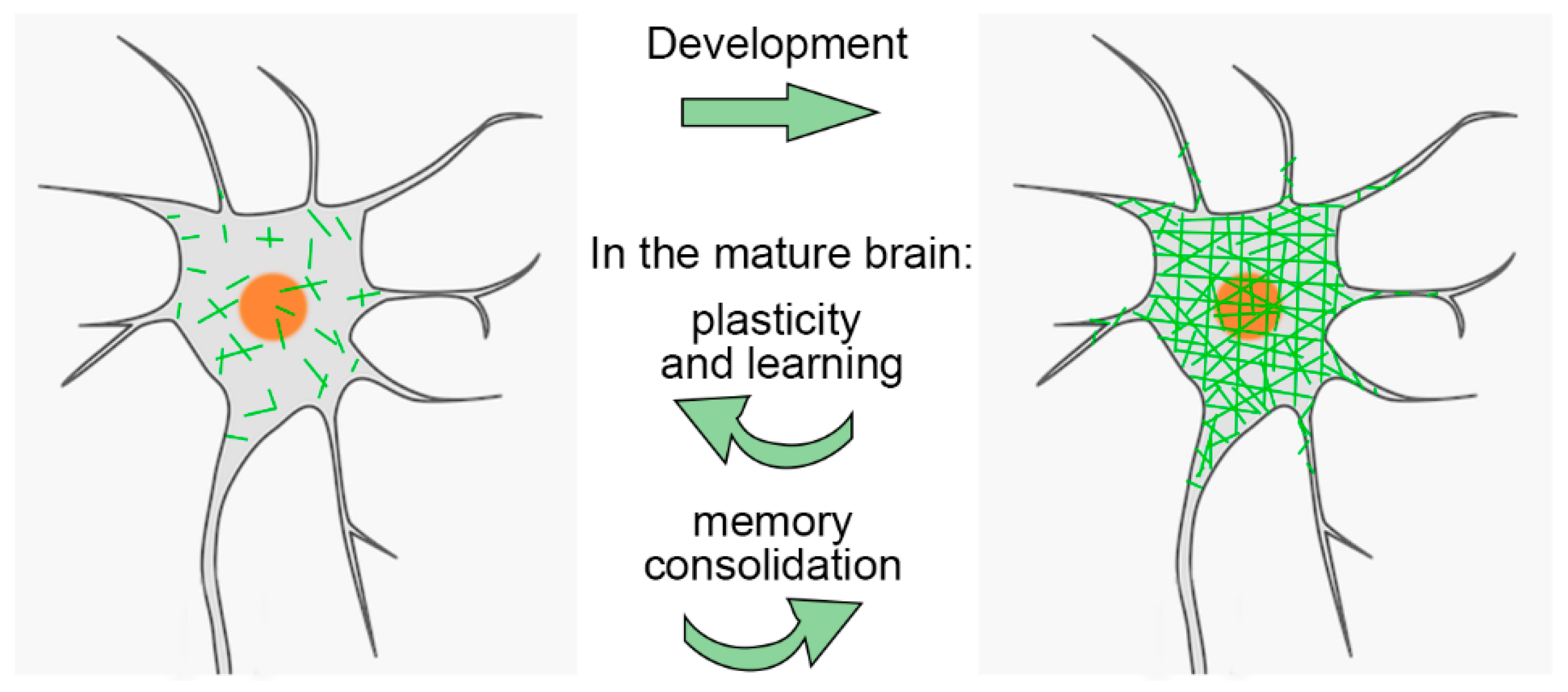 PNNs are especially important during brain development. They help close the "critical period" of heightened plasticity in childhood. Interestingly, while PNNs are degraded in adults, this plasticity can be partially restored. For example, PNN removal can promote recovery in stroke models
PNNs are especially important during brain development. They help close the "critical period" of heightened plasticity in childhood. Interestingly, while PNNs are degraded in adults, this plasticity can be partially restored. For example, PNN removal can promote recovery in stroke models This study investigates whether acupuncture has any impact on long-term health outcomes, such as mortality, disease progression, or complications, in individuals newly diagnosed with Parkinson’s disease (PD) in South Korea. Indeed, it is unclear what constitutes an effective acupuncture session for Parkinson's disease, and individuals interested should receive at least one session every two months. It is commonly believed that in PD, reduced mobility due to tremors, postural imbalance, and rigidity likely contributes to poor circulation and decreased gastrointestinal motility, leading to bowel obstructions and impaired swallowing, which can, in turn, result in recurrent aspiration pneumonia. The benefits may arise from the fact that people with PD might find it easier to move.
This study investigates whether acupuncture has any impact on long-term health outcomes, such as mortality, disease progression, or complications, in individuals newly diagnosed with Parkinson’s disease (PD) in South Korea. Indeed, it is unclear what constitutes an effective acupuncture session for Parkinson's disease, and individuals interested should receive at least one session every two months. It is commonly believed that in PD, reduced mobility due to tremors, postural imbalance, and rigidity likely contributes to poor circulation and decreased gastrointestinal motility, leading to bowel obstructions and impaired swallowing, which can, in turn, result in recurrent aspiration pneumonia. The benefits may arise from the fact that people with PD might find it easier to move.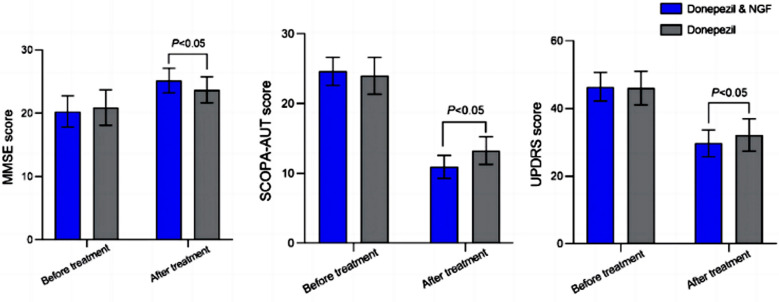 The authors focused on adiponectin, an adipocytokine, i.e. a molecule produced by adipose tissue, which is involved, among other things, in the regulation of lipid and glucose metabolism. Adiponectin modulates inflammatory cascades by modifying the action and production of inflammatory cytokines, but the link between adiponectin and Parkinson's disease is not obvious unless we consider that Parkinson's disease is due to a metabolic disorder. The relationship with the soluble tumor necrosis factor receptor (sTNFR) is even less obvious. Nothing in the article explains why these two molecules were studied.
The authors focused on adiponectin, an adipocytokine, i.e. a molecule produced by adipose tissue, which is involved, among other things, in the regulation of lipid and glucose metabolism. Adiponectin modulates inflammatory cascades by modifying the action and production of inflammatory cytokines, but the link between adiponectin and Parkinson's disease is not obvious unless we consider that Parkinson's disease is due to a metabolic disorder. The relationship with the soluble tumor necrosis factor receptor (sTNFR) is even less obvious. Nothing in the article explains why these two molecules were studied.