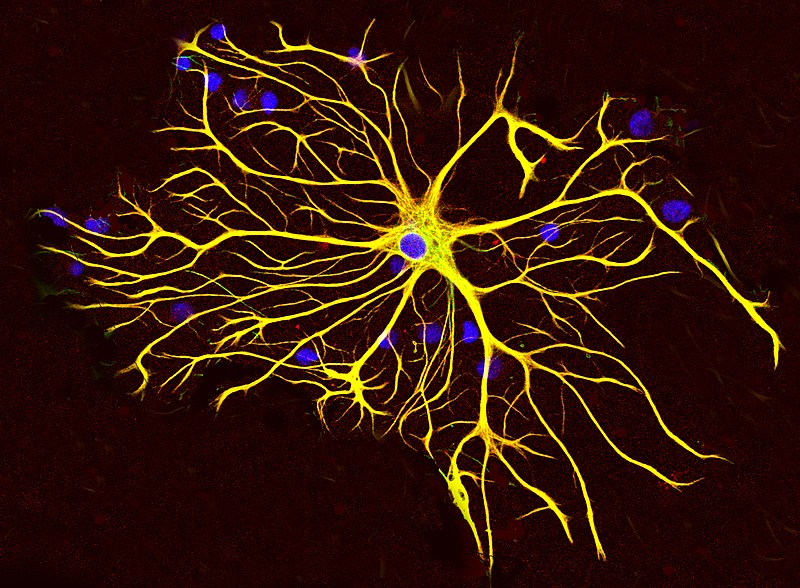
An article published by Aileen l. Pogue of Alchem Biotech Research in Canada and his colleagues at Louisiana State University discuss a pro-inflammatory toxin that may contribute to the development of Alzheimer's disease. The results are published in Frontiers in Neurology.
This article is (as usual) aggressively promoted, but probably not as new or important as its promoters would hope, nevertheless the subject is interesting even though it has made the subject of numerous scientific publications.
Intestinal dysbiosis has been implicated in the pathogenesis and progression of Alzheimer's disease by initiating and prolonging neuroinflammatory processes. Gut microbiota metabolites appear to be critical in the gut-brain axis mechanism. Gut microbiota metabolites, such as trimethylamine n-oxide, lipopolysaccharide, and short-chain fatty acids, are suggested to mediate systemic inflammation and intracerebral amyloidosis via endothelial dysfunction. New data suggest that the fungal microbiota may also influence the pathology of Alzheimer's disease.
Pogue and his colleagues believe they have found evidence that the lipopolysaccharide molecule in the human gastrointestinal (GI) tract generates an endotoxin which can perturb cells in the brain. Their paper links several recent observations linking lipopolysaccharide-induced increase in NF-kB signaling to increase in microRNA-30b.
NF-κB plays a key role in regulating the immune response to infection. Improper regulation of NF-κB has been linked to cancer, inflammatory and autoimmune diseases, septic shock, viral infection, and improper immune development. NF-κB has also been implicated in synaptic plasticity and memory processes.
MicroRNAs (miRNAs) are short non-coding RNAs that are involved in post-transcriptional regulation of gene expression by affecting both stability and translation of mRNAs. MicroRNAs are thought to have regulatory roles through complementarity with mRNA. These microRNAs regulate a number of genes associated with breast cancer.
The authors show that an increase in miRNA-30b is able to decrease the expression of neuron-specific neurofilament light chain (NFL) messenger RNA in stressed human neuronal-glial cells cultures.
Neurofilaments provide structural support to axons and regulate axon diameter, which influences nerve conduction velocity. Neurofilament light chain depletion therefore disrupts the normal shape of neuronal cells, their cytoarchitecture and synaptic organization. Neurofilament light chains are a useful marker for monitoring disease in amyotrophic lateral sclerosis, multiple sclerosis, Alzheimer's disease, and more recently Huntington's disease.
The presence of endotoxins, when detected in the blood, is called endotoxemia. Endotoxemia is associated with obesity, diet, cardiovascular disease and diabetes.
There is experimental and observational evidence that lipopolysaccharide may play a role in depression. Administration of lipopolysaccharide in mice can lead to depressive symptoms, and there appear to be elevated levels of lipopolysaccharide in some people with depression. Inflammation can sometimes play a role in the development of depression, and lipopolysaccharide is pro-inflammatory.
Finally, lipopolysaccharide-induced inflammation can induce cellular senescence, as has been demonstrated for lung epithelial cells and microglial cells (the latter leading to neurodegeneration).
In this study, researchers detail the pathway of BF-lipopolysaccharide from the gut to the brain and its mechanisms of action once there.
For them, BF-lipopolysaccharide leaks out of the gastrointestinal tract, crosses the blood-brain barrier via the circulatory system and gains access to the cerebral compartments. Next, it increases inflammation in brain cells and inhibits neuron-specific neurofilament light (NF-L), a protein that supports cellular integrity.
A deficiency of this protein leads to progressive atrophy of neuronal cells, and ultimately to cell death, as seen in neurons affected by Alzheimer's disease. They also report that an adequate intake of dietary fiber can prevent the process.
Indeed, 60% of gut microbiome variation is attributable to diet. Therefore, modulating the gut microbiome through dietary means could be an effective approach to reduce the risk of Alzheimer's disease.
Data from animal studies have suggested that dietary fat acts as the primary macronutrient responsible for postprandial endotoxemia, and that the quantity and quality of dietary fat differentially influence metabolic endotoxemia.
Additionally, healthy diets high in unsaturated fatty acids have been associated with lower circulating levels of lipopolysaccharide, which is strongly associated with lower pro-inflammatory markers. Conversely, consumption of diets high in energy or saturated fat has been associated with increased postprandial levels of lipopolysaccharide and increased circulating levels of pro-inflammatory markers.
Since people do not eat single nutrients and instead consume a diverse range of foods and a combination of nutrients that are likely to be interacting, studying the effects of whole diets offers the possibility of accounting for the interactions between different nutrients.
It is also probable that introducing variety in diet helps in having a diverse microbiome. Yet has people age, digestion becomes more difficult and aging people often prefer to not change their diet.
Thus, dietary habits may be more predictive of a real effect on the gut microbiome and Alzheimer's disease risk than foods or nutrients taken in isolation.
 Source: J.M.Garg - Own work, via Wikipedia
Source: J.M.Garg - Own work, via Wikipedia The compounds were further evaluated against Alzheimer's disease using a scopolamine-induced memory impairment mice model. It was observed that antioxidant and anticholinesterase properties of these compounds contribute significantly toward their remarkable potential to improve cognitive functioning.
The compounds were further evaluated against Alzheimer's disease using a scopolamine-induced memory impairment mice model. It was observed that antioxidant and anticholinesterase properties of these compounds contribute significantly toward their remarkable potential to improve cognitive functioning.
 Gynostemma pentaphyllum is used in folk medicine, typically as an herbal tea.
Gynostemma pentaphyllum is used in folk medicine, typically as an herbal tea. Le diabète sucré est considéré comme un facteur de risque pour de nombreuses maladies neurodégénératives, dont la maladie d'Alzheimer. Des études ont en effet montré le dysfonctionnement de la signalisation de l'insuline dans le cerveau, c'est à dire que les cellules du cerveau sont affamés par manque de glucose.
Le diabète sucré est considéré comme un facteur de risque pour de nombreuses maladies neurodégénératives, dont la maladie d'Alzheimer. Des études ont en effet montré le dysfonctionnement de la signalisation de l'insuline dans le cerveau, c'est à dire que les cellules du cerveau sont affamés par manque de glucose. Alzheimer's disease (Alzheimer's disease) is progressive brain disease that affects cognition, memory and behavior.
Alzheimer's disease (Alzheimer's disease) is progressive brain disease that affects cognition, memory and behavior.
 Three important discoveries showed how these mental maps were implemented in the mammalian brain.
* The first, in the early 1970s, is that hippocampal neurons, called place cells, respond to the position of the animal.
* The second, in the early 1990s, is that neurons in neighboring regions, called head direction cells, respond in the direction of the animal's head. This makes it possible to manipulate movement information and see how the location and lead direction cells react.
* The third finding was that the organization of neurons in the dorsomedial entorhinal cortex, named grid cells, closely resemble a sheet of squared paper organized in a hexagonal fashion and suggests that place cells can use grid cells to calculate distances.
Three important discoveries showed how these mental maps were implemented in the mammalian brain.
* The first, in the early 1970s, is that hippocampal neurons, called place cells, respond to the position of the animal.
* The second, in the early 1990s, is that neurons in neighboring regions, called head direction cells, respond in the direction of the animal's head. This makes it possible to manipulate movement information and see how the location and lead direction cells react.
* The third finding was that the organization of neurons in the dorsomedial entorhinal cortex, named grid cells, closely resemble a sheet of squared paper organized in a hexagonal fashion and suggests that place cells can use grid cells to calculate distances.
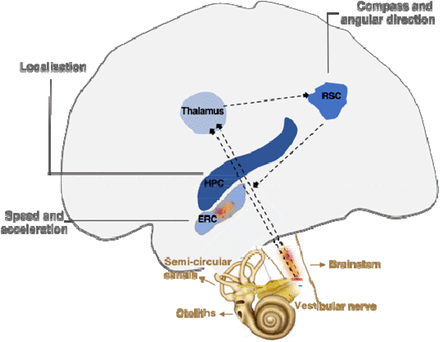 Deficits of path integration, ie of these mental maps, manifest themselves at the onset of Alzheimer's disease. Decades before the expected onset of the disease subtle changes in pathway integration are also present in adults at genetic risk for Alzheimer's disease.
Deficits of path integration, ie of these mental maps, manifest themselves at the onset of Alzheimer's disease. Decades before the expected onset of the disease subtle changes in pathway integration are also present in adults at genetic risk for Alzheimer's disease.