Un article semble démontrer de façon inattendue que certains médicaments contre le VIH et l'hépatite pourraient contribuer à réduire le risque de maladie d'Alzheimer. La maladie d'Alzheimer (MA) reste l'une des maladies les plus complexes de la médecine moderne, tant en termes de traitement que de prévention. Mais des recherches par une équipe de l'université de Virginie menée par Joseph Magagnoli et Meenakshi Ambati, pointent vers un allié potentiel inattendu : une classe de médicaments antiviraux initialement développés pour traiter le VIH et l'hépatite B.
On sait que réduire l'inflammation induite par NLRP3 peut réduire l'impact de la maladie d'Alzheimer. Les inhibiteurs nucléosidiques de la transcriptase inverse (NRTI), sont approuvés par la Food and Drug Administration (FDA) des États-Unis pour traiter les infections par le virus de l'immunodéficience humaine (VIH) et l'hépatite B. L'équipe de Joseph Magagnoli et Meenakshi Ambati avait montré antérieurement des NRTI inhibent également l'activation de l'inflammasome indépendamment de leur activité antirétrovirale. D'autres groupes ont par la suite confirmé cette observation.
Cependant il s'agit là des études pré-cliniques, il y en a des dizaines de milliers chaque année pour la maladie d'Alzheimer et elles ne suffisent pas à convaincre un investisseur de financer une étude clinique qui en phase III coûte au minimum une vingtaine de millions de Dollars. Il faut donc des indications qu'un tel médicament pourrait bénéficier à de spatients humains. Plusieurs moyens peuvent être employés, mais si l'on veut prouver qu'un médicament déjà commercialisé est utile dans une autre maladie, il est intéressant d'étudier les bases de données historiques qui analyse l'évolution d'une cohorte sur une longue période.
Les chercheurs ont analysé deux importantes bases de données de santé américaines afin de répondre à une question simple, mais essentielle : les personnes prenant des inhibiteurs nucléosidiques de la transcriptase inverse (NRTI), un groupe de médicaments antiviraux, présentent-elles un risque moindre de développer la maladie d'Alzheimer ?
Les chercheurs ont examiné plus de 270 000 patients dans deux bases de données : - Administration de la santé des anciens combattants (VA) – 72 193 patients entre 2000 et 2024 - MarketScan (assurance privée) – 199 005 patients entre 2006 et 2020
L’étude a porté sur des personnes âgées de 50 ans ou plus, atteintes du VIH ou d’hépatite B, mais sans diagnostic préalable de maladie d’Alzheimer. L’étude a permis de déterminer les personnes à qui des NRTI étaient prescrits et la durée de leur prise.
L’analyse a pris en compte un large éventail de facteurs susceptibles d’influencer le risque de maladie d’Alzheimer, notamment l’âge, le sexe, l’origine ethnique, les maladies cardiaques, le diabète, la dépression, les lésions cérébrales, etc. Les chercheurs ont également réalisé une analyse spécifique (appelée appariement par score de propension) afin de s’assurer de comparer des groupes de personnes similaires : celles qui prenaient des NRTI et celles qui n’en prenaient pas.
 Ils ont confirmé qu'une exposition prolongée aux NRTI était associée à un risque moindre de développer la maladie d’Alzheimer.
- D’après les données du VA, chaque année supplémentaire de traitement par NRTI était associée à une réduction de 4 à 6 % du risque de maladie d’Alzheimer.
- D’après les données de MarketScan, la réduction était encore plus marquée : de 10 à 13 % par année d’utilisation.
Ils ont confirmé qu'une exposition prolongée aux NRTI était associée à un risque moindre de développer la maladie d’Alzheimer.
- D’après les données du VA, chaque année supplémentaire de traitement par NRTI était associée à une réduction de 4 à 6 % du risque de maladie d’Alzheimer.
- D’après les données de MarketScan, la réduction était encore plus marquée : de 10 à 13 % par année d’utilisation.
Au total il semblerait que la prise d’un NRTI soit associée à une réduction de 32 à 37 % du risque de maladie d’Alzheimer. Attention il s'agit bien de réduction du rique de développer cette maladie, il ne s'agit pas d'un médicament qui guérirait de cette maladie. Une question importante est alors (si ce médicament est autorisé) sur quels critères devrait-on prescrire ce médicament à des personnes apparement saines?
Il s’agit d’une étude observationnelle et non d’un essai clinique ; elle ne prouve donc pas que les NRTI préviennent la maladie d’Alzheimer. Mais la concordance des résultats obtenus sur deux grandes populations, avec des ajustements importants pour d'autres facteurs de santé, rend ces conclusions remarquables.
Ce qui est particulièrement intriguant, c'est que les NRTI sont déjà approuvés et utilisés, ce qui signifie qu'ils pourraient être réutilisés plus rapidement que des médicaments entièrement nouveaux. Et comme la maladie d'Alzheimer est une maladie aux options thérapeutiques limitées, même de petites avancées en matière de prévention pourraient faire une différence significative.
Des essais contrôlés randomisés, sont nécessaires pour confirmer si ces médicaments ont réellement un effet protecteur contre la maladie d'Alzheimer et si ce bénéfice l'emporte sur les risques, en particulier chez les personnes non atteintes du VIH ou de l'hépatite B. Néanmoins, cette recherche ouvre une voie prometteuse.
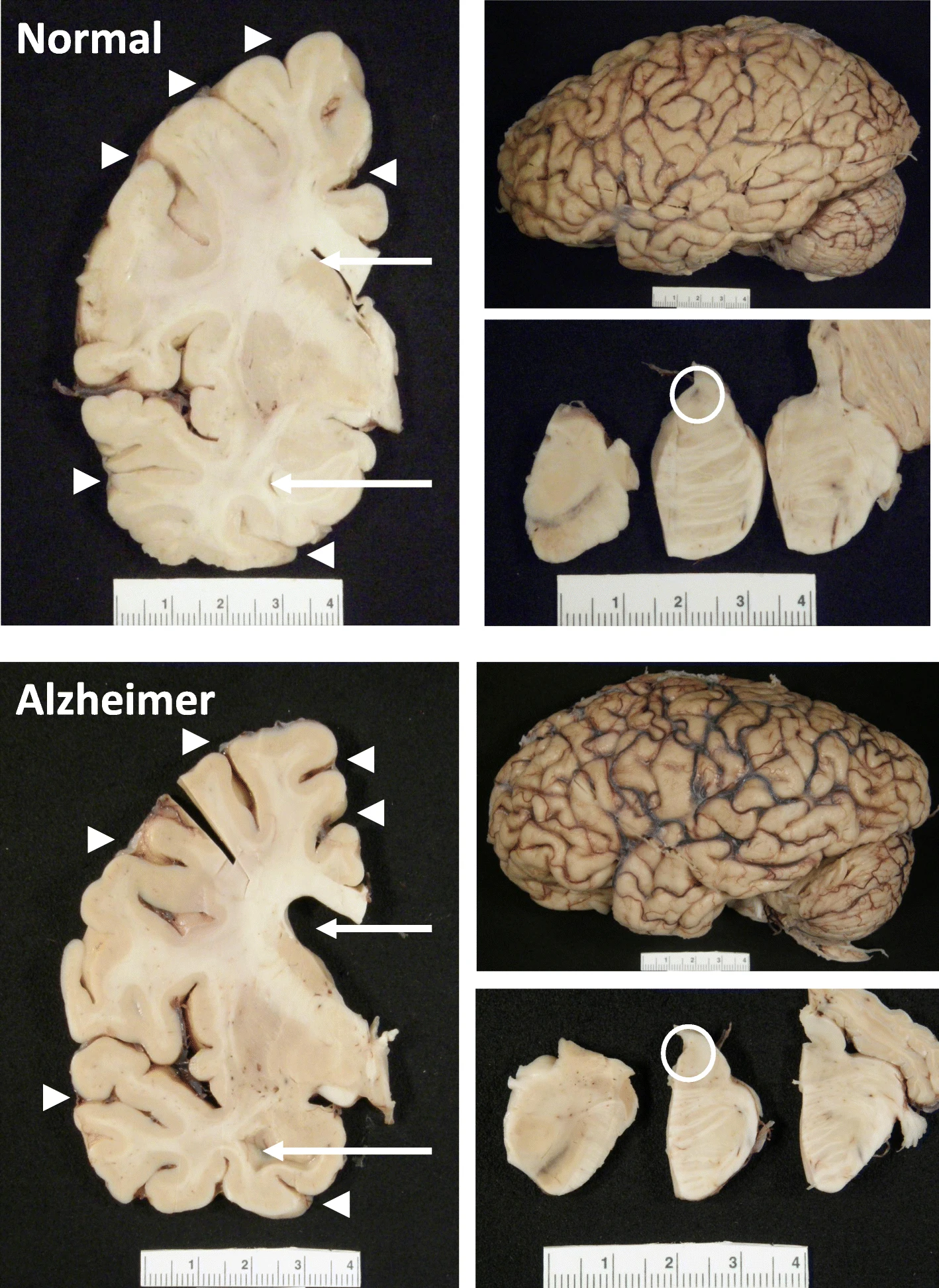 The hippocampus, a small, seahorse-shaped structure buried deep within the brain, is best known for its role in memory formation and learning. It is an exceptionally vulnerable structure, with perfusion deficits often observed in diseases related to learning and memory. However, a brain affected by Alzheimer's disease tends to exhibit at least moderate cortical atrophy, including in the precuneus and posterior cingulate gyrus. It should be noted that the posterior cingulate gyrus is adjacent to the hippocampus.
The hippocampus, a small, seahorse-shaped structure buried deep within the brain, is best known for its role in memory formation and learning. It is an exceptionally vulnerable structure, with perfusion deficits often observed in diseases related to learning and memory. However, a brain affected by Alzheimer's disease tends to exhibit at least moderate cortical atrophy, including in the precuneus and posterior cingulate gyrus. It should be noted that the posterior cingulate gyrus is adjacent to the hippocampus.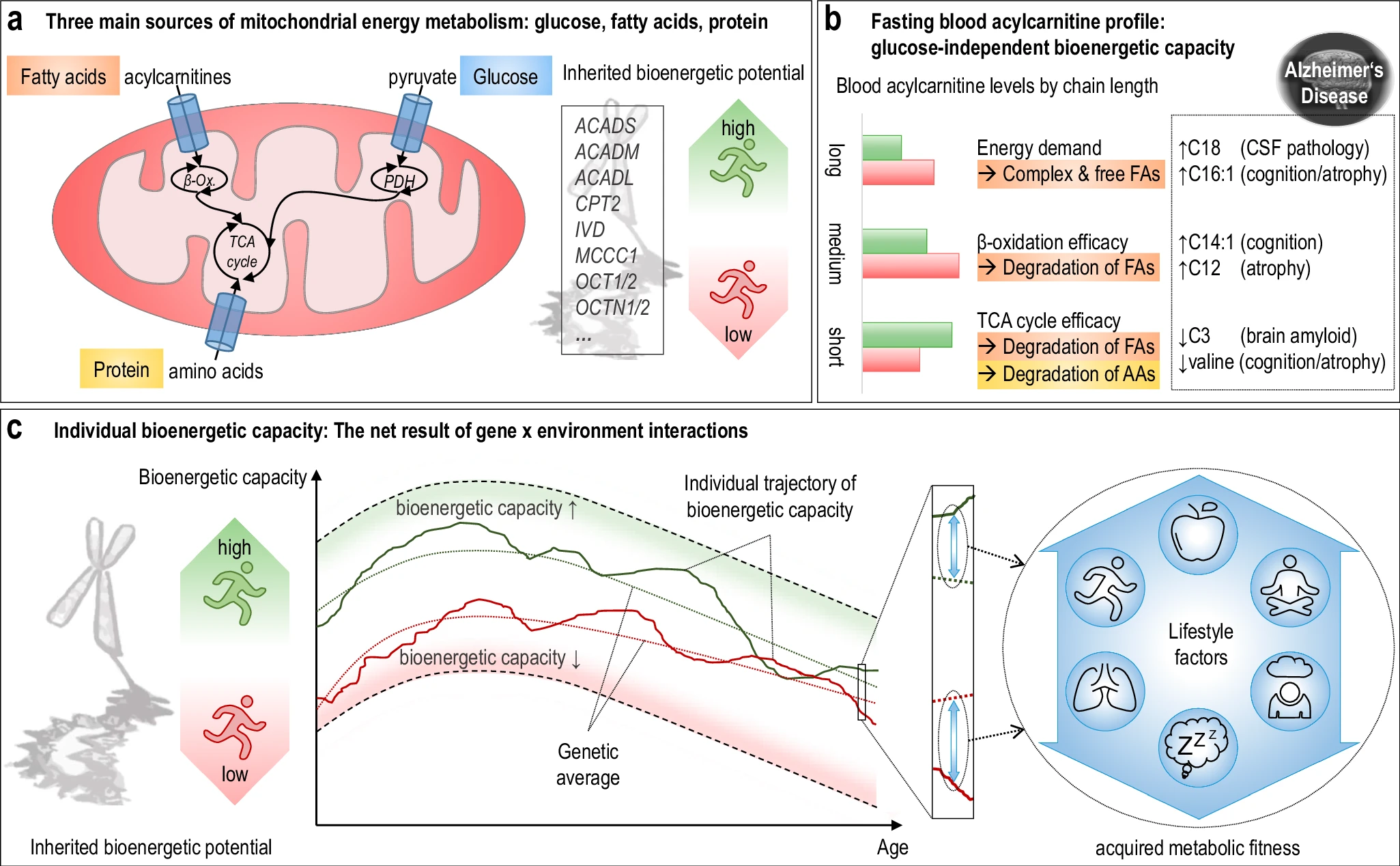 The researchers used acylcarnitine profiles from blood samples to identify distinct bioenergetic subgroups in Alzheimer's Disease (AD) patients and evaluate how bioenergetic capacity relates to disease progression.
They used data from 1,531 participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI), and identified several bioenergetic subgroups with significant differences in AD biomarkers, cognitive function, and brain glucose metabolism.
These subgroups were primarily determined by modifiable factors (40-60%) related to beta-oxidation function, rather than genetic factors, suggesting potential for intervention.
The researchers used acylcarnitine profiles from blood samples to identify distinct bioenergetic subgroups in Alzheimer's Disease (AD) patients and evaluate how bioenergetic capacity relates to disease progression.
They used data from 1,531 participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI), and identified several bioenergetic subgroups with significant differences in AD biomarkers, cognitive function, and brain glucose metabolism.
These subgroups were primarily determined by modifiable factors (40-60%) related to beta-oxidation function, rather than genetic factors, suggesting potential for intervention. Alzheimer's disease research has produced many hypotheses over the years, including cholinergic, inflammatory, viral, mitochondrial, tau, and amyloid. However, none of these hypotheses have led to treatments that can stop or reverse the disease. This leads to a search for new theories to explain these failures. But this may be because interventions occur too late in the disease progression, with brain damage irreparable and compensatory mechanisms saturated.
Alzheimer's disease research has produced many hypotheses over the years, including cholinergic, inflammatory, viral, mitochondrial, tau, and amyloid. However, none of these hypotheses have led to treatments that can stop or reverse the disease. This leads to a search for new theories to explain these failures. But this may be because interventions occur too late in the disease progression, with brain damage irreparable and compensatory mechanisms saturated.
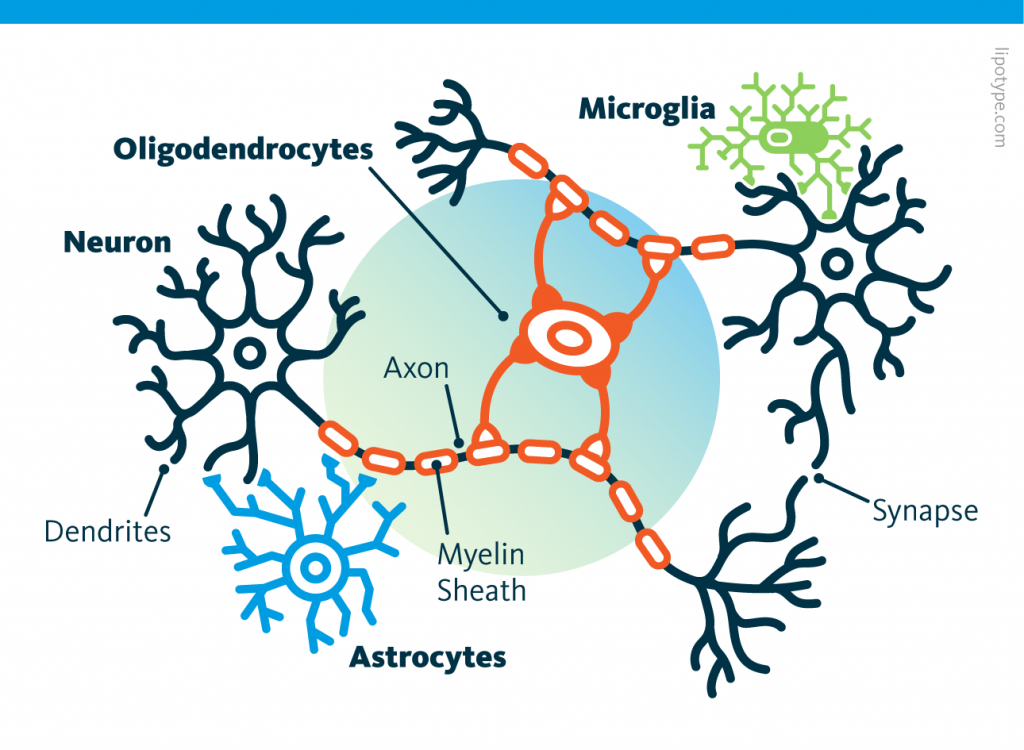 Curiously, scientists are rather looking to directly convert astrocytes into neurons, despite the enormous morphological difference between these two types of cells.
Curiously, scientists are rather looking to directly convert astrocytes into neurons, despite the enormous morphological difference between these two types of cells. Those other cells, which compose half of the brain's cells, are receiving more attention. There are multiple types but normally they are there to assist neurons in their task. A simplified view tells that neurons are a sort of plumbing system and the glial cells are the real actors in the brain.
Those other cells, which compose half of the brain's cells, are receiving more attention. There are multiple types but normally they are there to assist neurons in their task. A simplified view tells that neurons are a sort of plumbing system and the glial cells are the real actors in the brain. Source: Nephron via Wikipedia
Source: Nephron via Wikipedia
 Source: Peta
Source: Peta Running before learning aids in the formation of new memories, yet, running after learning promotes the forgetting of recently acquired information!
Running before learning aids in the formation of new memories, yet, running after learning promotes the forgetting of recently acquired information!