Huntington’s Disease and C9orf72 ALS: Shared Mechanisms and Therapeutic Hopes
Approximately 70,000 people have been diagnosed with Huntington’s disease (HD) in the U.S. and Europe, with hundreds of thousands more at risk of inheriting the condition. Despite the clear genetic cause of HD, there are currently no approved therapies that delay onset or slow progression.
Both Huntington's disease and C9orf72-linked ALS, while clinically distinct, share a common hallmark: long, abnormal repetitions of DNA bases. The success of antisense oligonucleotides (ASOs) in spinal muscular atrophy (SMA, SMN1 gene) in 2017, followed by gene therapy in 2019, gave researchers confidence to pursue similar strategies in HD and C9orf72 ALS. Progress in treating one of these repeat expansion diseases may provide hope for others.
1. Genetic Basis
1.1 Huntington’s disease (HD)
HD is caused by an expanded CAG trinucleotide repeat in the HTT gene. - Normal alleles: up to approximately 26 repeats - Pathogenic threshold: 36 or more repeats
CAG encodes glutamine, leading to a mutant protein with an expanded polyglutamine (polyQ) tract. This toxic protein disrupts neuronal function and accumulates throughout the body, contributing not only to neurodegeneration but also to systemic issues like muscle atrophy, cardiac problems, impaired glucose tolerance, weight loss, osteoporosis, and testicular atrophy.

1.2 C9orf72 ALS/FTD
C9orf72-related ALS and frontotemporal dementia (FTD) are caused by an expanded GGGGCC (G4C2) hexanucleotide repeat in the C9orf72 gene. - Normal alleles: up to approximately 30 repeats - Pathogenic alleles: hundreds to thousands
The expansion causes disease through several mechanisms: - Reduced C9orf72 protein levels - Formation of toxic RNA foci - Production of abnormal dipeptide repeat proteins via repeat-associated non-ATG (RAN) translation
1.3 Other repeat expansion diseases
- Spinocerebellar ataxias (SCAs) – many caused by CAG expansions
- Fragile X syndrome – CGG expansion in FMR1
- Myotonic dystrophy – CTG expansion in DMPK
2. Therapeutic Approaches: Shared Strategies
2.1 Antisense oligonucleotides (ASOs)
ASOs aim to reduce toxic transcripts. - HD: ASOs targeting HTT mRNA have reached clinical trials (e.g., Roche/Ionis). - C9orf72 ALS: ASOs targeting repeat-containing transcripts are in early-stage trials.
2.2 Gene silencing/editing
The most advanced approach in HD is uniQure’s AMT-130 gene therapy: - Uses an AAV vector to deliver microRNAs designed to silence mutant HTT. - Administered through MRI-guided stereotactic neurosurgery directly into the striatum. - Clinical trials (U.S. and Europe) are ongoing, with promising early results showing up to 75% slowing in disease progression in high-dose patients over 36 months.
These approaches are not yet cures, but they show that disease modification is possible. Advances in vector design (AAVs, lipid nanoparticles) are directly transferable to other repeat expansion disorders.
2.3 Targeting RNA structures
Small molecules that bind abnormal RNA structures (hairpins, G-quadruplexes) are under development for C9orf72 ALS and myotonic dystrophy, with potential extension to CAG-repeat disorders like HD.
2.4 Modulating protein homeostasis
Strategies to boost autophagy, proteasome activity, or molecular chaperones could reduce toxic protein aggregates in both HD and C9orf72 ALS.
3. Translating Progress Across Diseases
Research tools—such as assays for RNA foci, protein aggregation, and repeat instability—are shared across laboratories working on different repeat expansion disorders. Breakthroughs in one disease can therefore be rapidly tested in others.
Delivery challenges are also common: therapies must reach neurons in the brain and spinal cord. Advances in intrathecal ASO delivery or viral vector engineering benefit all disorders in this family.
In summary: Huntington’s disease and C9orf72 ALS/FTD are distinct conditions, but they share a unifying principle: DNA repeat expansions that disrupt RNA and protein homeostasis. Therapeutic strategies—including antisense oligonucleotides, RNA-targeting drugs, and gene-editing technologies—are broadly applicable across these diseases. Progress in one field accelerates progress in others, offering shared hope for patients facing these devastating neurodegenerative disorders.
 The authors found that key immune signaling proteins, specifically those containing Death Fold Domains (DFDs) (like ASC, FADD, BCL10, MAVS, TRADD), exist in a unique physical state inside our cells called metastable supersaturation. These full-length adaptors retain nucleation barriers and are able to exist supersaturated in cells. In contrast, many receptors and effectors do not. This localizes the “spring-loaded” behaviour to central adaptors that link receptor sensing to downstream cell-fate decisions.
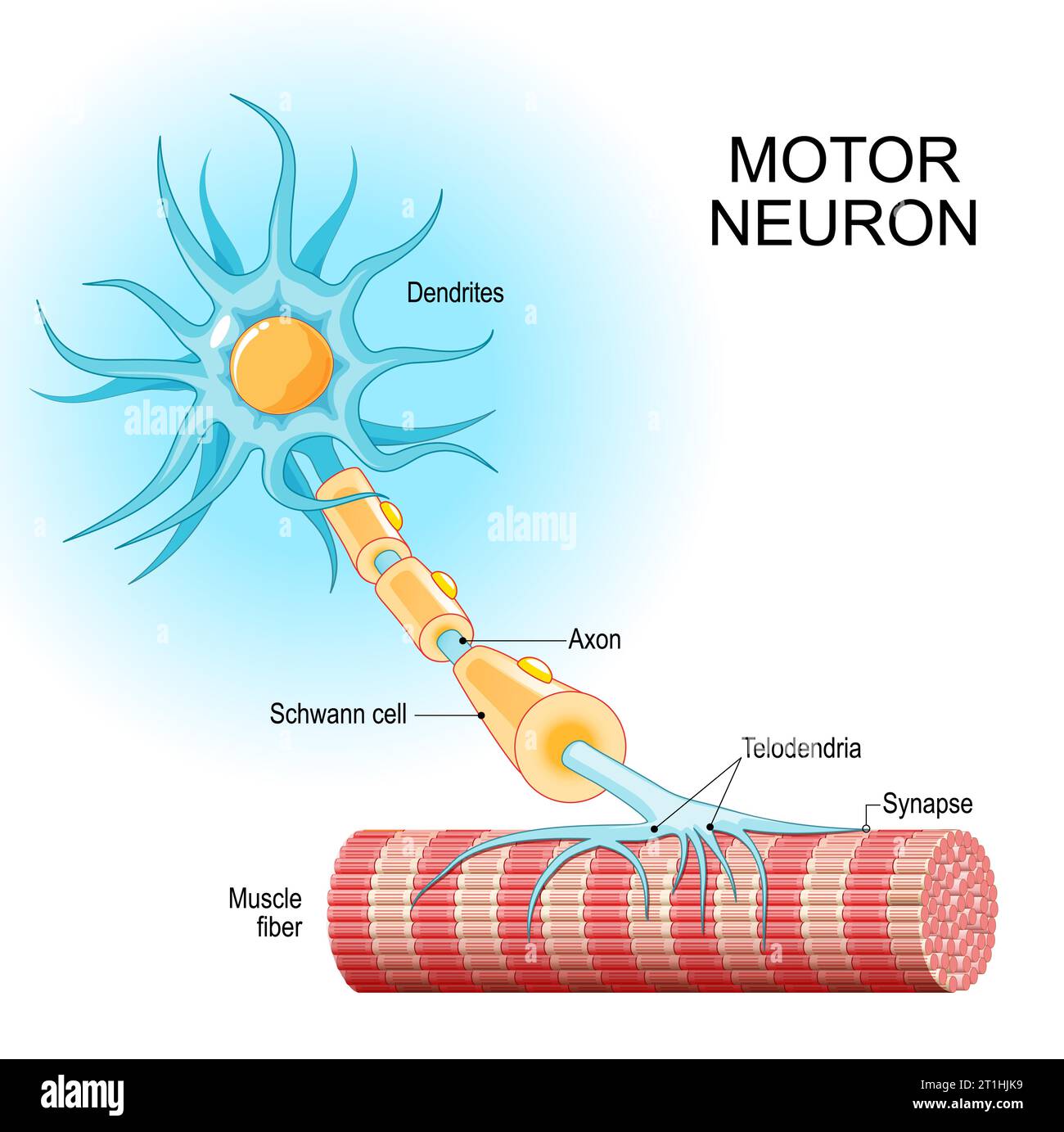
The authors found that key immune signaling proteins, specifically those containing Death Fold Domains (DFDs) (like ASC, FADD, BCL10, MAVS, TRADD), exist in a unique physical state inside our cells called metastable supersaturation. These full-length adaptors retain nucleation barriers and are able to exist supersaturated in cells. In contrast, many receptors and effectors do not. This localizes the “spring-loaded” behaviour to central adaptors that link receptor sensing to downstream cell-fate decisions.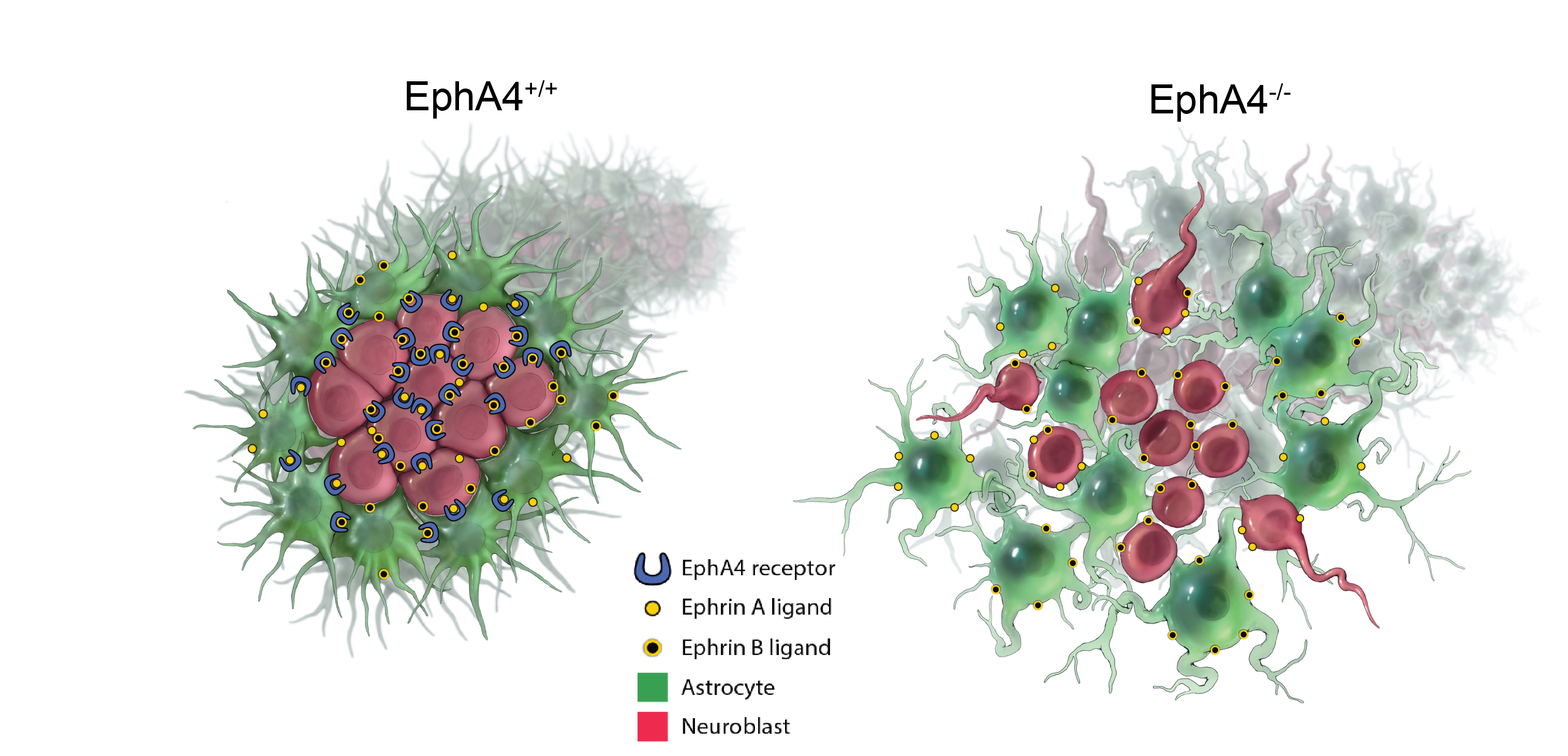 At this stage, neuroblasts express key transcription factors like ISL1 and LHX3, which establish the fundamental identity of the motor neuron. The neuroblast begins to resemble more to a motor neuron: They extend a long axon out of the spinal cord towards their target muscle. The cell also starts to acquire its specific electrical properties. Then the neuron reaches its target muscle, forms a neuromuscular junction, and becomes a fully functional, electrically active cell. At this point, the early master regulators like ISL1 and LHX3 are largely downregulated, and the neuron enters its final, mature state.
At this stage, neuroblasts express key transcription factors like ISL1 and LHX3, which establish the fundamental identity of the motor neuron. The neuroblast begins to resemble more to a motor neuron: They extend a long axon out of the spinal cord towards their target muscle. The cell also starts to acquire its specific electrical properties. Then the neuron reaches its target muscle, forms a neuromuscular junction, and becomes a fully functional, electrically active cell. At this point, the early master regulators like ISL1 and LHX3 are largely downregulated, and the neuron enters its final, mature state.
 The authors designed a genetic therapy with an AAV virus vector to make mature neurons express two proteins that are only expressed in the immature state.
The AAVs were specifically engineered to target motor neurons. In the study conducted on mice, the administration mode of the AAV viral vector was able to specifically infect the spinal motor neurons.
Once inside the mature motor neurons, the AAV released the therapeutic genes. This caused the neurons to begin expressing ISL1 and LHX3 again
By re-expressing ISL1 and LHX3, the researchers essentially re-activate that original "immature" genetic program. This causes the mature neuron to revert to a state that is genetically and functionally similar to its younger self, with renewed resilience and stress-coping abilities.
They believe that turning on the immature genetic program essentially re-awakens the neuron's dormant ability to regrow and repair itself. While mature neurons in the central nervous system have very limited regenerative capacity, the authors are suggesting that ISL1 and LHX3 could be flipping a switch that bypasses this limitation.
The authors designed a genetic therapy with an AAV virus vector to make mature neurons express two proteins that are only expressed in the immature state.
The AAVs were specifically engineered to target motor neurons. In the study conducted on mice, the administration mode of the AAV viral vector was able to specifically infect the spinal motor neurons.
Once inside the mature motor neurons, the AAV released the therapeutic genes. This caused the neurons to begin expressing ISL1 and LHX3 again
By re-expressing ISL1 and LHX3, the researchers essentially re-activate that original "immature" genetic program. This causes the mature neuron to revert to a state that is genetically and functionally similar to its younger self, with renewed resilience and stress-coping abilities.
They believe that turning on the immature genetic program essentially re-awakens the neuron's dormant ability to regrow and repair itself. While mature neurons in the central nervous system have very limited regenerative capacity, the authors are suggesting that ISL1 and LHX3 could be flipping a switch that bypasses this limitation. De façon similaire, la force des souris modifiés génétiquement est nettement plus basse au début du traitement qu’à la fin, c’est l’inverse de ce qu’on pourrait attendre d’une souris qui serait de plus en plus affaiblie.
Et pourquoi ce groupe de souris aurait-il une force plus faible au début de l’expérience ? S’il n’y a pas de sélection à priori, les différents groupes de souris (traités et non traités) devraient avoir la même force.
De façon similaire, la force des souris modifiés génétiquement est nettement plus basse au début du traitement qu’à la fin, c’est l’inverse de ce qu’on pourrait attendre d’une souris qui serait de plus en plus affaiblie.
Et pourquoi ce groupe de souris aurait-il une force plus faible au début de l’expérience ? S’il n’y a pas de sélection à priori, les différents groupes de souris (traités et non traités) devraient avoir la même force. What should we consider about all this? Maybe we should ask why scientists are searching for new drugs instead of focusing on compounds of drugs that have already shown some effects. Perhaps everyone wants to get rich, so they avoid exploring drugs that can't be patented.
What should we consider about all this? Maybe we should ask why scientists are searching for new drugs instead of focusing on compounds of drugs that have already shown some effects. Perhaps everyone wants to get rich, so they avoid exploring drugs that can't be patented. Les progrès réalisés dans la chimie médicinale des ASO ont amélioré leur profil pharmacodynamique, permettant ainsi une administration sûre et efficace au système nerveux central. Les thérapies ASO pour la SLA se sont rapidement développées au cours des deux dernières décennies, et les ASO ciblant SOD1, C9orf72 et ATXN2 sont actuellement en essais cliniques pour les formes familiales ou sporadiques de SLA.
Les progrès réalisés dans la chimie médicinale des ASO ont amélioré leur profil pharmacodynamique, permettant ainsi une administration sûre et efficace au système nerveux central. Les thérapies ASO pour la SLA se sont rapidement développées au cours des deux dernières décennies, et les ASO ciblant SOD1, C9orf72 et ATXN2 sont actuellement en essais cliniques pour les formes familiales ou sporadiques de SLA. Identifié par criblage in vitro, l'ASO ION363 développé par la société IONIS qui a aussi développé Tofersen, cible le 6e intron de FUS (SLA avec une mutation P525L).
ION363 réduit les taux de protéines de liaison à l'ARN insolubles et insolubles associées aux agrégats, telles que hnRNPA1 et ralentit la neurodégénérescence des motoneurones lombaires et la perte d'innervation de la jonction neuromusculaire.
Identifié par criblage in vitro, l'ASO ION363 développé par la société IONIS qui a aussi développé Tofersen, cible le 6e intron de FUS (SLA avec une mutation P525L).
ION363 réduit les taux de protéines de liaison à l'ARN insolubles et insolubles associées aux agrégats, telles que hnRNPA1 et ralentit la neurodégénérescence des motoneurones lombaires et la perte d'innervation de la jonction neuromusculaire. The researchers specifically focused on molecules that could reduce or reverse stress granule formation, particularly those that act directly on stress granule proteins and may be useful as therapeutic agents. From the initial screening, lipoamide emerged as a novel, potent modulator of stress granules.
Once lipoamide was identified as a hit, the researchers sought to determine its effects in cells regarding specificity, potency, intracellular localization, and its effects on other cellular condensates.
The researchers specifically focused on molecules that could reduce or reverse stress granule formation, particularly those that act directly on stress granule proteins and may be useful as therapeutic agents. From the initial screening, lipoamide emerged as a novel, potent modulator of stress granules.
Once lipoamide was identified as a hit, the researchers sought to determine its effects in cells regarding specificity, potency, intracellular localization, and its effects on other cellular condensates.