For once, we are going to talk about an article about Huntington's disease. Huntington's disease (sometimes called Huntington's chorea) is a rare hereditary disease that results in neurological degeneration causing significant motor, cognitive, and psychiatric disorders, and progressing to the loss of autonomy and then death. This disease occurs in adults aged between 35 and 50 on average. The progression of the disease follows a rhythm and a form that varies greatly from one individual to another.
The genetic abnormality that causes Huntington's disease is a greater than normal increase (20 times) of the repetition of three nucleotides (C, A, and G - called the CAG codon or triplet) within the HTT gene encoding the huntingtin protein. This blog focuses on three neurodegenerative diseases, ALS (sometimes called Charcot's disease or Lou Gehrig's disease), Parkinson's disease, or Alzheimer's disease. But in fact, it is only in books that these diseases are clearly categorized, in reality, we distinguish multiple subtypes to each of these categorizations and moreover, these diseases share characteristics both between themselves and with other neurodegenerative diseases.
A variant of ALS (C9orf72) closely resembles Huntington's disease in some aspects because it too is characterized by repeat expansions of a gene, C9orf72. Repeat expansions of the C9orf72 gene are responsible for about 40% of genetic ALS and 25% of genetic FTD.
The article we are discussing today focuses on the genetic and molecular mechanisms underlying Huntington's disease (HD), with a particular emphasis on the dynamics of CAG repeat expansions in the huntingtin (HTT) gene.
It explores the implications for the pathogenesis and therapeutics of HD. Although ALS is not cited by the authors, this paper has implications for that disease as well as for about 30 others.
An expansion of CAG triplet causes Huntington's disease repeats in the HTT gene. Normal alleles have 15 to 30 CAGs; disease-causing alleles have 36 or more. Longer CAG repeats correlate with earlier onset.
Scientists identified the progression of CAG repeat expansion with age by analyzing somatic mosaicism, or the variability in CAG repeat lengths between different cells within an individual. This variability increases over time, and the progression of CAG repeat length has been linked to cell-specific processes that cause expansions as individuals age.
The authors found that HTT alleles are not inherently toxic, but become harmful after somatic expansion exceeds approximately 150 repeats.
This toxicity results in asynchronous and rapid neuronal decline, challenging the understanding of Huntington’s disease as a slowly progressive disease.
 They measured somatic repeat expansion over time in individual neurons from donors of different ages. They found that early-phase expansions (e.g., from 40 to 80 CAG repeats) were slow and stochastic, taking decades, while later expansions (e.g., from 80 to 150 repeats) occurred more rapidly.
They then analyzed genetic markers and DNA repair mechanisms associated with repeat instability, such as those involving DNA mismatch repair (MMR) proteins (e.g., MSH3, PMS1). Variants in these genes have been shown to influence the rate of somatic instability. The progression of CAG repeat expansion is driven by errors in DNA replication, repair, and maintenance, particularly in neurons. Key mechanisms include:
They measured somatic repeat expansion over time in individual neurons from donors of different ages. They found that early-phase expansions (e.g., from 40 to 80 CAG repeats) were slow and stochastic, taking decades, while later expansions (e.g., from 80 to 150 repeats) occurred more rapidly.
They then analyzed genetic markers and DNA repair mechanisms associated with repeat instability, such as those involving DNA mismatch repair (MMR) proteins (e.g., MSH3, PMS1). Variants in these genes have been shown to influence the rate of somatic instability. The progression of CAG repeat expansion is driven by errors in DNA replication, repair, and maintenance, particularly in neurons. Key mechanisms include:
Striatal projection neurons, the cells most affected by Huntington’s disease, exhibit higher levels of somatic instability than other cell types. This may be due to their unique transcriptional and metabolic profiles, which make them more susceptible to DNA damage and inefficient repair.
Expansion dynamics have been categorized into phases: - Phase A (36–80 repeats): Slow and stochastic over decades. - Phase B (80–150 repeats): Faster and more predictable. - Phase C (>150 repeats): Gene expression changes begin, leading to cellular dysfunction. Toxicity threshold: Neurons derepress other genes (e.g., CDKN2A/B) and eventually die.
These findings highlight how somatic mosaicism and age-related repeat expansions are central to the pathogenesis of Huntington’s disease and provide a framework for understanding similar processes in other repeat expansion disorders.
The somatic expansion mechanism may extend to other repeat expansion disorders, such as myotonic dystrophy or some forms of spinocerebellar ataxia.
The C9orf72 variant in ALS involves a hexanucleotide repeat expansion (GGGGCC) in a noncoding gene region. Although distinct from the CAG repeats in the Huntington’s disease coding region, there are notable parallels. As in Huntington’s disease, somatic instability of the repeat expansion has been observed in ALS C9orf72. The degree of mosaicism may influence disease onset and progression. C9orf72 expansions lead to toxic RNA foci and dipeptide repeat (DPR) proteins, leading to neuronal dysfunction and death. This reflects Huntington’s disease idea that toxicity only occurs after substantial repeat expansion. In ALS, motor neurons are selectively vulnerable. As in Huntington’s disease, the specific cell types affected may result from unique somatic instability dynamics.
Therapeutic potential: This work in Huntington’s disease suggests that modulation of DNA repair pathways (e.g., MSH3) may stabilize nucleotide repeats, thereby delaying toxicity.
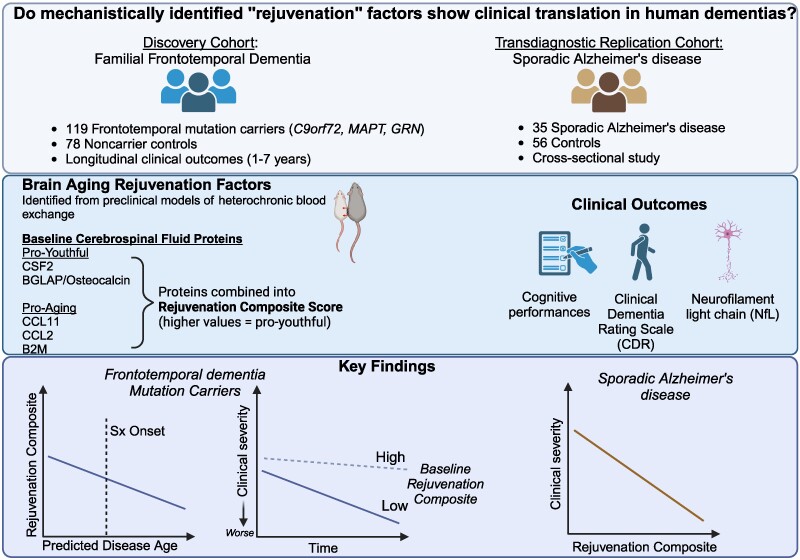 The results appear relatively reliable because the scientists found similar effects in two different types of dementia. The effects were seen across multiple measures (cognitive, functional, and biological markers).
The results appear relatively reliable because the scientists found similar effects in two different types of dementia. The effects were seen across multiple measures (cognitive, functional, and biological markers). The endoplasmic reticulum (ER) is an important organelle in cells that is involved in protein conformation. This step occurs after protein synthesis by ribosomes and after conformation, the new protein will be sent to its final destination by the Golgi apparatus. Protein conformation requires energy, so when disease occurs, the ER may not be able to properly conform the new proteins.
The endoplasmic reticulum (ER) is an important organelle in cells that is involved in protein conformation. This step occurs after protein synthesis by ribosomes and after conformation, the new protein will be sent to its final destination by the Golgi apparatus. Protein conformation requires energy, so when disease occurs, the ER may not be able to properly conform the new proteins. Throughout the study, incidence rates ranged from 2/100,000 person-years in 2008 to 2.77/100,000 in 2021), with the latter year being the year with the highest incidence recorded.
Throughout the study, incidence rates ranged from 2/100,000 person-years in 2008 to 2.77/100,000 in 2021), with the latter year being the year with the highest incidence recorded. Although there is a large literature on the split hand phenomenon in ALS, knowledge remains limited for other motor neuron diseases, including SMA.
Although there is a large literature on the split hand phenomenon in ALS, knowledge remains limited for other motor neuron diseases, including SMA. Dinitrophenol acts as a proton transporter in the mitochondrial membrane, inhibiting oxidative phosphorylation of ATP and making energy production less efficient. This is because some of the energy that is normally produced from cellular respiration is wasted as heat. This inefficiency is proportional to the dose of dinitrophenol that is absorbed. Thus, as the dose increases, energy production becomes less efficient: metabolism is then activated - more fat is burned - to compensate for the inefficiency and meet energy demands.
Dinitrophenol acts as a proton transporter in the mitochondrial membrane, inhibiting oxidative phosphorylation of ATP and making energy production less efficient. This is because some of the energy that is normally produced from cellular respiration is wasted as heat. This inefficiency is proportional to the dose of dinitrophenol that is absorbed. Thus, as the dose increases, energy production becomes less efficient: metabolism is then activated - more fat is burned - to compensate for the inefficiency and meet energy demands. Angel Bu is the first author, while Ritu Raman is the senior author; the other authors are from MIT’s Department of Mechanical Engineering and MIT’s Koch Institute for Integrative Cancer Research. The authors matured a set of motor neurons on a gel that was a kind of carpet into which they embedded tiny magnets. They then used an external magnet to shake the carpet—and the neurons—back and forth. In this way, they made the neurons work, for 30 minutes a day.
Angel Bu is the first author, while Ritu Raman is the senior author; the other authors are from MIT’s Department of Mechanical Engineering and MIT’s Koch Institute for Integrative Cancer Research. The authors matured a set of motor neurons on a gel that was a kind of carpet into which they embedded tiny magnets. They then used an external magnet to shake the carpet—and the neurons—back and forth. In this way, they made the neurons work, for 30 minutes a day.