Abnormal protein aggregation within cells is a recurring phenomenon in Parkinson's disease (PD), Alzheimer's disease (AD), and amyotrophic lateral sclerosis (ALS). Current approaches use antibodies to target these aggregates, but this is a rudimentary approach, as little is known about the causes of their formation, or whether they are the cause or consequence of the disease.
Cells are an incredibly crowded environment, and their molecules undergo Brownian motion, which thwarts their biological function. Making the cell less dense and more soluble would certainly alleviate some molecular problems. There are various approaches, including those that use phase transitions.
Recent research sheds surprising light on the dynamic relationship between mitochondrial activity, ATP levels, and neuronal cytoplasmic fluidity, all of which play a critical role in controlling protein aggregation.
The researchers used mouse giant goblet cell cultures to analyze presynaptic viscosity using real-time confocal microscopy. These cells are characterized by large glutamatergic nerve terminals, ideally suited for real-time imaging. Rather than focusing on individual proteins, the team took a holistic approach, using a technique called fluorescence recovery after photobleaching (FRAP) of soluble green fluorescent protein (cGFP) to assess the overall viscosity of the axonal cytosol.
Cytosolic viscosity can reflect the extent of protein aggregation; Greater aggregation means less free diffusion of cGFP, indicating a more "solidified" cytosol.
Synapses are hotspots for mitochondria, which provide the ATP needed for neurotransmission. By labeling active mitochondria and comparing their location to cGFP mobility, the study revealed that regions with greater mitochondrial activity exhibited higher cytosolic fluidity. This suggests a direct link between ATP production and the maintenance of a more soluble and functional presynaptic environment.
To further investigate this, the team inhibited mitochondrial function using FCCP and other mitochondrial blockers. As ATP production decreased, cGFP diffusion decreased sharply, suggesting that the cytosol was becoming more viscous due to protein aggregation. It is important to note that this effect was specific to mitochondrial inhibition: blocking glycolysis had little effect.
Even components of the synaptic release mechanism, such as synaptic vesicles (SVs) and active zones (AZs), exhibited reduced mobility under mitochondrial stress, reinforcing the idea that energy depletion disrupts the fluid phase of the cytoplasm.
To test whether ATP could restore the altered cytosol state, the researchers administered ATP directly to neurons. They found that ATP not only restored cGFP diffusion but also reduced the size and number of protein aggregates. To test whether enhancing endogenous ATP production could mitigate the protein aggregation linked to mitochondrial dysfunction, the researchers turned to NMN, a molecule known to boost NAD⁺ levels and support mitochondrial health.
They treated neurons with NMN and observed the following key outcomes:
Partial restoration of cytoplasmic fluidity: In neurons with compromised mitochondrial activity (such as those derived from PARK2 or TDP-43 mutant patients), NMN treatment significantly improved the diffusion of soluble proteins like cGFP. While not as dramatic as direct ATP infusion, NMN nonetheless reduced cytosolic viscosity.
Reduction in aggregate burden: In both mouse neurons under mitochondrial stress and hiPSC-derived human neurons from neurodegenerative disease patients, NMN treatment lowered the accumulation of insoluble protein aggregates.
Improved ATP levels: NMN supplementation helped increase intracellular ATP concentrations, presumably by enhancing mitochondrial NAD⁺-dependent enzymatic activity, which supports oxidative phosphorylation.
These results suggest that NMN supports the same protective pathway as ATP, but indirectly, by restoring mitochondrial capacity to generate ATP and maintain a more fluid intracellular environment.
The mechanism appears to be biophysical rather than biochemical: ATP acts as a hydrotrope, a molecule that keeps other proteins dissolved and prevents them from forming aggregates.
The researchers then examined whether this principle held true for specific proteins involved in neurodegenerative diseases, including:
α-synuclein (mutant SNCA and SNCA-A53T), PARK2 – Parkinson's disease
APP, Amyloid, Tau – Alzheimer's disease
TDP-43 – ALS
These purified proteins were able to undergo liquid-liquid phase separation (LPS) and form condensates in vitro. ATP was able to dissolve many of these condensates in a concentration-dependent manner, although mutant or misfolded versions (e.g., SNCA-A53T) required higher ATP concentrations to dissolve.
When the aggregates were left to incubate for longer, some (notably SNCA-A53T) began to form protofibrils, elongated, fibril-like structures similar to those observed in real-life pathology. Here again, ATP could reverse this phenomenon, but with reduced efficiency.
Even under crowded conditions (mirrored by the addition of PEG), ATP retained some ability to prevent or dissolve aggregates, although the effect was less potent.
The team then studied neurons derived from Human induced pluripotent stem cells (hiPSCs) from patients with Parkinson's disease (PARK2 mutation) and ALS (TDP-43 mutation). These neurons exhibited reduced cytosolic fluidity, lower ATP levels, and greater protein aggregation than healthy controls.
This supports the idea that ATP deficiency and mitochondrial dysfunction contribute to the condensation of pathogenic proteins in human neurodegenerative diseases.
Implications for Drug Development
This research redefines our approach to therapeutic targets in neurodegenerative diseases. Instead of seeking to eliminate aggregates after their formation, we could:
Target mitochondrial function to preserve ATP production at synapses.
Use small molecules that mimic the hydrotropic effects of ATP to maintain cytoplasmic fluidity.
Develop drugs that prevent the formation of LPS (lipoproteinases) of key disease proteins by improving their solubility.
ATP itself is not a drug molecule in the traditional sense, but these results open new avenues for small molecules capable of acting like ATP to maintain protein solubility or prevent aggregate formation at an early stage.
Conclusion
Neurodegenerative diseases are often viewed from a genetic or protein perspective, but this study provides a biophysical perspective: the physical state of the cytosol itself is crucial. If cells cannot maintain a fluid and soluble environment, primarily due to energy deficiency, aggregation may become inevitable.
This is not just about treating symptoms or even eliminating aggregates afterward. It is about preserving the cellular environment so that neurons can withstand stress and maintain their function. As the field continues to explore how biophysical properties such as viscosity, solubility, and phase separation interact with disease, the role of ATP may prove central, not only as a fuel, but also as a key regulator of neuronal health.
 What should we consider about all this? Maybe we should ask why scientists are searching for new drugs instead of focusing on compounds of drugs that have already shown some effects. Perhaps everyone wants to get rich, so they avoid exploring drugs that can't be patented.
What should we consider about all this? Maybe we should ask why scientists are searching for new drugs instead of focusing on compounds of drugs that have already shown some effects. Perhaps everyone wants to get rich, so they avoid exploring drugs that can't be patented.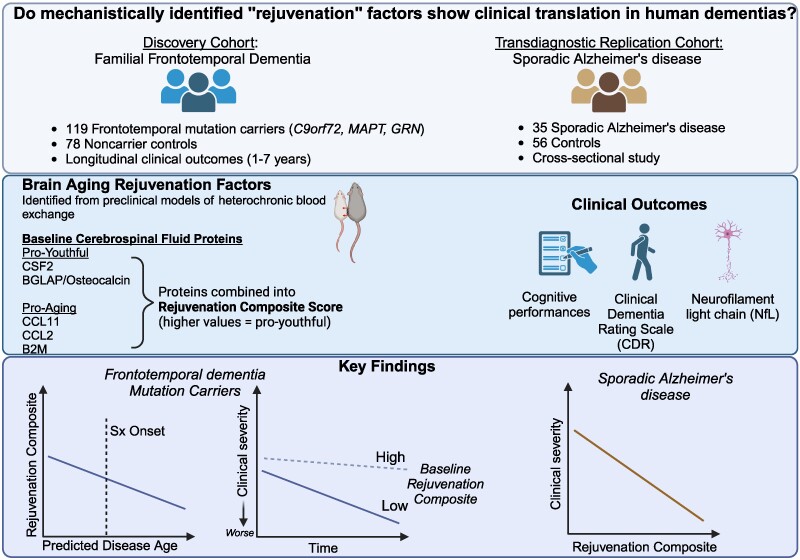 The results appear relatively reliable because the scientists found similar effects in two different types of dementia. The effects were seen across multiple measures (cognitive, functional, and biological markers).
The results appear relatively reliable because the scientists found similar effects in two different types of dementia. The effects were seen across multiple measures (cognitive, functional, and biological markers).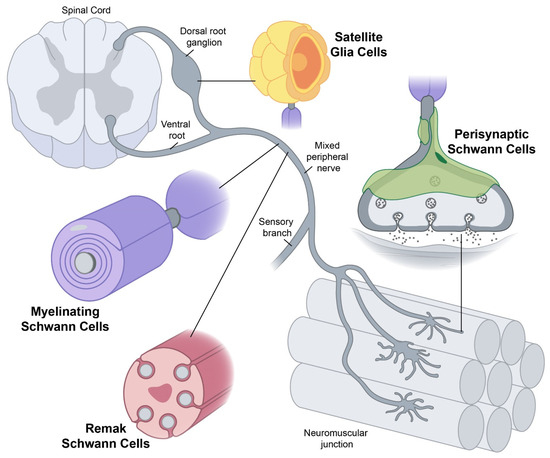 Yet conceptually associating Schwann cells and ALS is not common, ALS is a disease of the central nervous system (upper motor neurons in the brain and spine) while Schwann cells are located in the peripheral nervous system (lower motor neurons with their bodies from the spine and terminating in muscles). This does not mean there are no relations between the two types of cells, and it's a common view now that neurons are not independent, self-sufficient entities and that they are cared for by a large number of other cell types.
Yet conceptually associating Schwann cells and ALS is not common, ALS is a disease of the central nervous system (upper motor neurons in the brain and spine) while Schwann cells are located in the peripheral nervous system (lower motor neurons with their bodies from the spine and terminating in muscles). This does not mean there are no relations between the two types of cells, and it's a common view now that neurons are not independent, self-sufficient entities and that they are cared for by a large number of other cell types.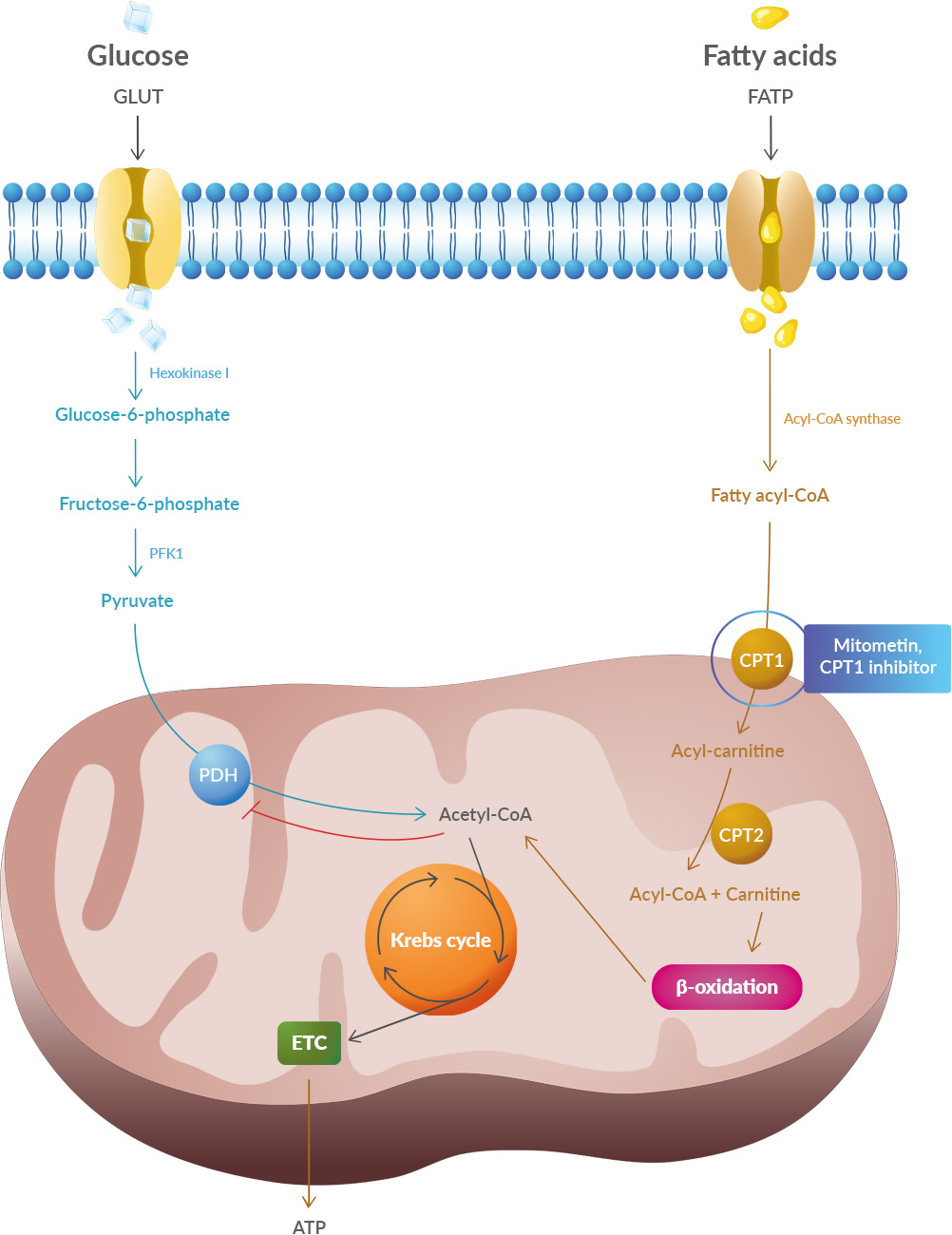 Studies have shown that oxidative stress and endoplasmic reticulum stress are correlated and can lead to protein misfolding (Abramov et al., 2020). Accumulation of misfolded proteins causes cellular damage and mitochondrial dysfunction and is associated with a range of neurodegenerative diseases, including ALS (misfolded SOD1, TDP-43, C9orf72) (McAlary et al., 2020), Parkinson's disease (misfolded α-synuclein) and Alzheimer disease (misfolded Aβ and Tau) (Abramov et al., 2020).
Studies have shown that oxidative stress and endoplasmic reticulum stress are correlated and can lead to protein misfolding (Abramov et al., 2020). Accumulation of misfolded proteins causes cellular damage and mitochondrial dysfunction and is associated with a range of neurodegenerative diseases, including ALS (misfolded SOD1, TDP-43, C9orf72) (McAlary et al., 2020), Parkinson's disease (misfolded α-synuclein) and Alzheimer disease (misfolded Aβ and Tau) (Abramov et al., 2020).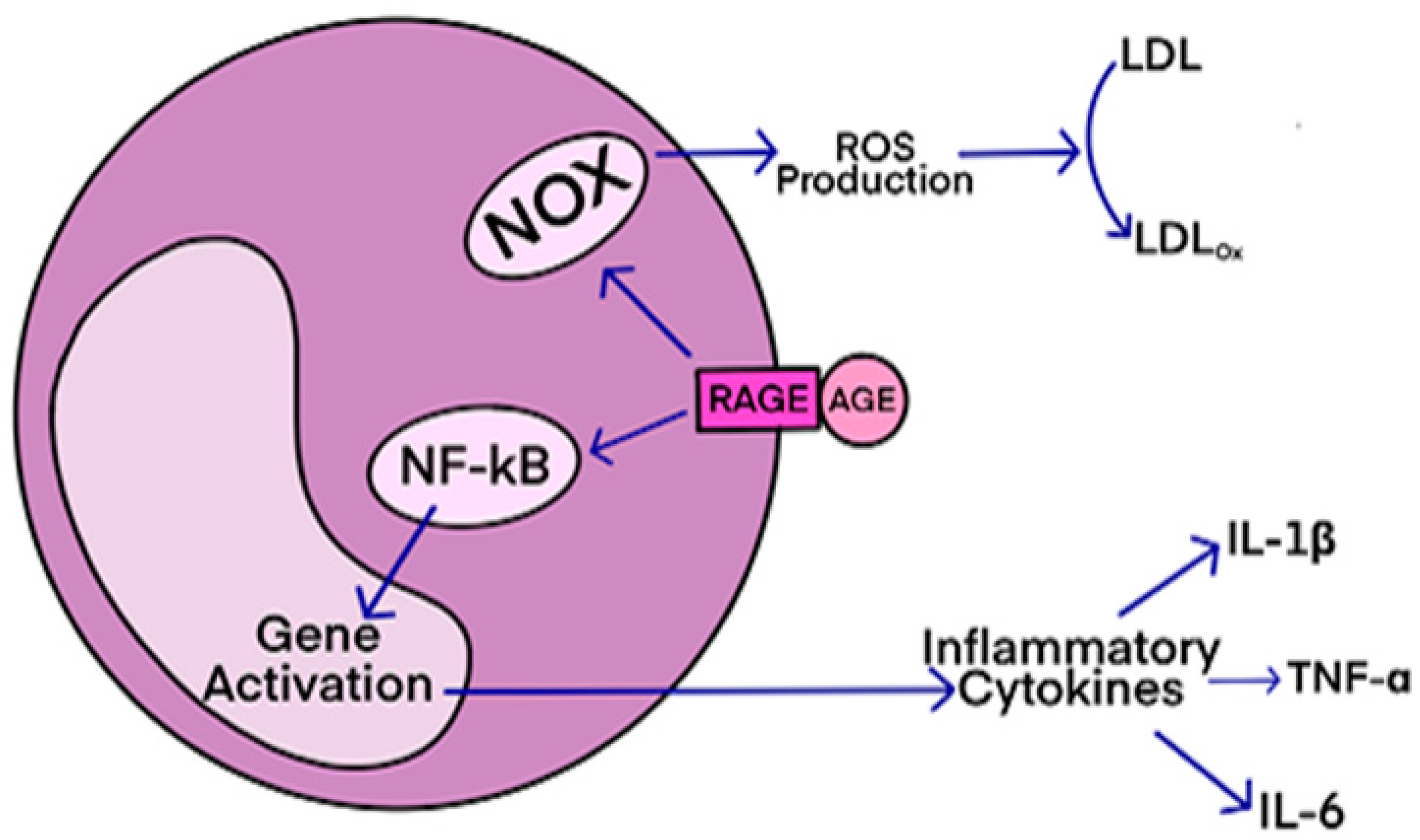 AGEs have diverse structures, but only a limited number have been characterized. Some AGEs are small molecules formed through proteolytic degradation of protein-crosslinked or protein-modified AGEs. Imbalance between the formation and destruction of AGEs, particularly under conditions of oxidative stress, results in excessive accumulation and disease progression.
AGEs have diverse structures, but only a limited number have been characterized. Some AGEs are small molecules formed through proteolytic degradation of protein-crosslinked or protein-modified AGEs. Imbalance between the formation and destruction of AGEs, particularly under conditions of oxidative stress, results in excessive accumulation and disease progression.
