A recent study analyzed the plasma proteomes of over 2,000 participants to identify proteins and biological pathways associated with Alzheimer's disease and related disorders. https://www.nature.com/articles/s43587-025-00965-4
The widely held hypothesis among scientists is that sticky amyloid plaques in the brain are a hallmark of Alzheimer's disease. With the announcement that the initial work on this hypothesis was fraudulent, along with hundreds of unsuccessful clinical trials, a growing number of scientists suggest that other processes must be at play.
This new study suggests that factors outside the brain, such as processes in the blood and other organs, may contribute to the disease. The authors show that several biological processes, including those related to the extracellular matrix, proteostasis, the immune system, and metabolism, play an important role in Alzheimer's disease. This means that what happens in the rest of the body could influence the brain and how quickly Alzheimer's disease progresses. The study also highlights the strong influence of the APOE ε4 genotype and lipoprotein biology.
Extracellular matrix (ECM):
The ECM is closely linked to cerebral β-amyloid (Aβ) deposition and cognitive decline. Some ECM proteins, such as SMOC1 and SPON1, are elevated in both plasma and the brains of patients with Alzheimer’s, while others, like HTRA1, show opposite trends in the two compartments. Changes in the ECM could impact cognitive function independently of β-amyloid buildup and may be connected to vascular integrity loss.
ECM proteins are found throughout the body and provide structural and biochemical support to cells and tissues. They are produced by various cell types, with fibroblasts being the most common source in connective tissues. Other cells, such as cartilage chondrocytes and kidney mesangial cells, also produce ECM components.
Proteostasis:
This process, involving protein synthesis and clearance, is linked to both Aβ plaques and cognitive function. Increased protein synthesis correlates with better cognition, while enhanced protein degradation links to poorer cognitive performance. Further research is needed to understand how these processes interact in the brain and peripheral tissues.
The proteostasis network, which manages protein synthesis, folding, and degradation, operates across multiple cell compartments in all tissues. As a result, proteins involved in this process originate from organs like the liver, muscle, and adipose tissue.
Immune system:
Activation of the immune response in the blood is strongly associated with declines in cognitive function, even after accounting for Aβ plaques. This suggests that peripheral immune responses may contribute significantly to cognitive impairment.
Proteins involved in immune processes are primarily produced by immune cells like leukocytes (white blood cells) and by other cells in immune-related organs such as the bone marrow, spleen, and thymus.
Synaptic proteins:
The synaptic protein NPTXR was the only protein consistently associated with cognitive performance across all examined groups; higher NPTXR levels correlated with better cognition. However, the study found mixed associations for other neuronal proteins—some indicated healthy brain function, while others signaled neuronal damage.
Metabolism:
Unlike in the brain and cerebrospinal fluid, where increased metabolic proteins associate with cognitive decline, plasma proteins related to metabolism show the opposite trend. This inverse relationship suggests that these proteins may originate from peripheral non-neuronal sources or that their transport across the blood-brain barrier is tightly regulated.
Metabolic proteins participate in processes like glycolysis and energy production. They are produced by cells in tissues such as skeletal muscle, fat, and the liver, which is central to metabolic regulation.
Lipoproteins and APOE ε4:
Lipoprotein biology is closely linked to Aβ buildup in the brain and cognitive function. Lower plasma levels of lipoprotein proteins, including APOE, associate with higher brain Aβ levels. The study also confirms the widespread effects of the APOE ε4 genotype, influencing multiple pathways such as cell division and microtubule functions, potentially connecting Aβ and tau pathologies.
The liver mainly produces and regulates lipoproteins like VLDL and HDL, which are crucial for lipid transport in the bloodstream.
Conclusion:
The study showed that some biological pathways, including the extracellular matrix, are similar across blood, cerebrospinal fluid, and brain, but others, like metabolism and synaptic pathways, differ significantly. These findings emphasize the importance of studying proteins in multiple body compartments to fully understand their role in Alzheimer’s.
The researchers note several limitations, such as incomplete data on factors like medication use, the absence of long-term follow-up data, and limited information on neurofibrillary tangle (NFT) burden in most groups.
In summary, the study illustrates that the complex processes underlying Alzheimer's disease can be detected in blood plasma, identifying potential targets for future therapies and biomarkers. It also supports the idea of using blood tests as a less invasive, more accessible way to study and monitor the disease progression.

 By Manu5 - http://www.scientificanimations.com/wiki-images/
By Manu5 - http://www.scientificanimations.com/wiki-images/ Ils ont confirmé qu'une exposition prolongée aux NRTI était associée à un risque moindre de développer la maladie d’Alzheimer.
- D’après les données du VA, chaque année supplémentaire de traitement par NRTI était associée à une réduction de 4 à 6 % du risque de maladie d’Alzheimer.
- D’après les données de MarketScan, la réduction était encore plus marquée : de 10 à 13 % par année d’utilisation.
Ils ont confirmé qu'une exposition prolongée aux NRTI était associée à un risque moindre de développer la maladie d’Alzheimer.
- D’après les données du VA, chaque année supplémentaire de traitement par NRTI était associée à une réduction de 4 à 6 % du risque de maladie d’Alzheimer.
- D’après les données de MarketScan, la réduction était encore plus marquée : de 10 à 13 % par année d’utilisation.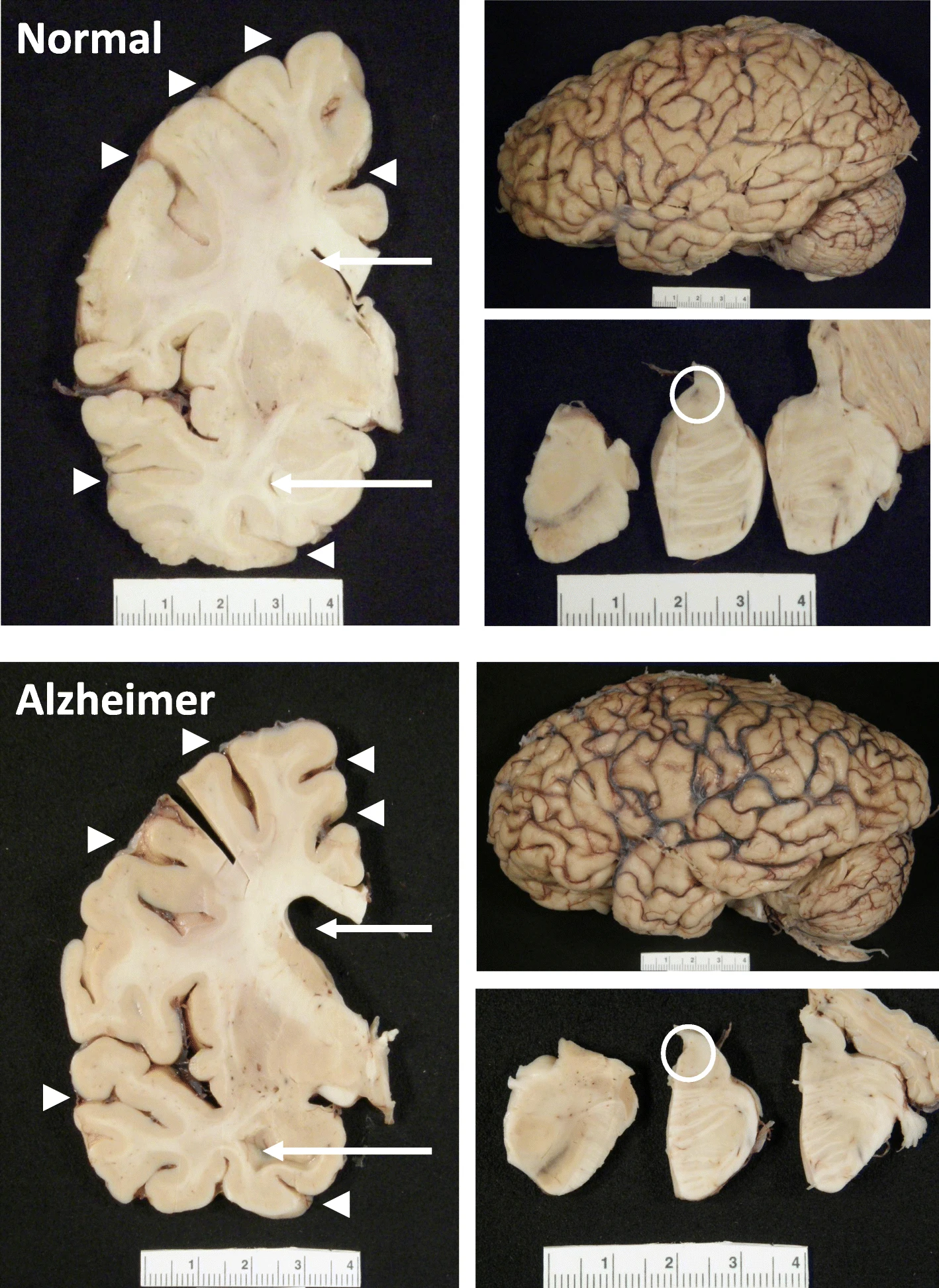 The hippocampus, a small, seahorse-shaped structure buried deep within the brain, is best known for its role in memory formation and learning. It is an exceptionally vulnerable structure, with perfusion deficits often observed in diseases related to learning and memory. However, a brain affected by Alzheimer's disease tends to exhibit at least moderate cortical atrophy, including in the precuneus and posterior cingulate gyrus. It should be noted that the posterior cingulate gyrus is adjacent to the hippocampus.
The hippocampus, a small, seahorse-shaped structure buried deep within the brain, is best known for its role in memory formation and learning. It is an exceptionally vulnerable structure, with perfusion deficits often observed in diseases related to learning and memory. However, a brain affected by Alzheimer's disease tends to exhibit at least moderate cortical atrophy, including in the precuneus and posterior cingulate gyrus. It should be noted that the posterior cingulate gyrus is adjacent to the hippocampus.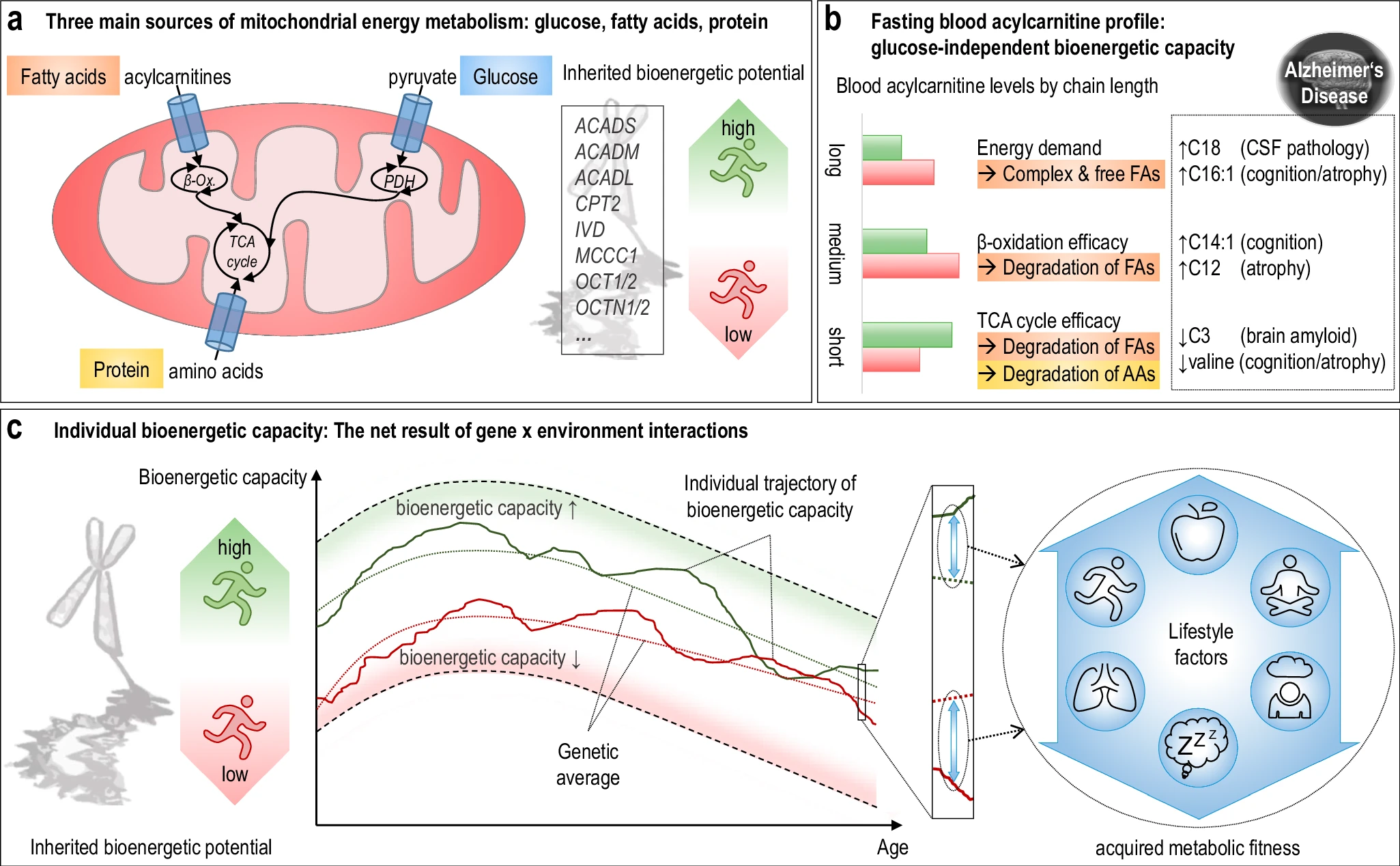 The researchers used acylcarnitine profiles from blood samples to identify distinct bioenergetic subgroups in Alzheimer's Disease (AD) patients and evaluate how bioenergetic capacity relates to disease progression.
They used data from 1,531 participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI), and identified several bioenergetic subgroups with significant differences in AD biomarkers, cognitive function, and brain glucose metabolism.
These subgroups were primarily determined by modifiable factors (40-60%) related to beta-oxidation function, rather than genetic factors, suggesting potential for intervention.
The researchers used acylcarnitine profiles from blood samples to identify distinct bioenergetic subgroups in Alzheimer's Disease (AD) patients and evaluate how bioenergetic capacity relates to disease progression.
They used data from 1,531 participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI), and identified several bioenergetic subgroups with significant differences in AD biomarkers, cognitive function, and brain glucose metabolism.
These subgroups were primarily determined by modifiable factors (40-60%) related to beta-oxidation function, rather than genetic factors, suggesting potential for intervention. Alzheimer's disease research has produced many hypotheses over the years, including cholinergic, inflammatory, viral, mitochondrial, tau, and amyloid. However, none of these hypotheses have led to treatments that can stop or reverse the disease. This leads to a search for new theories to explain these failures. But this may be because interventions occur too late in the disease progression, with brain damage irreparable and compensatory mechanisms saturated.
Alzheimer's disease research has produced many hypotheses over the years, including cholinergic, inflammatory, viral, mitochondrial, tau, and amyloid. However, none of these hypotheses have led to treatments that can stop or reverse the disease. This leads to a search for new theories to explain these failures. But this may be because interventions occur too late in the disease progression, with brain damage irreparable and compensatory mechanisms saturated.
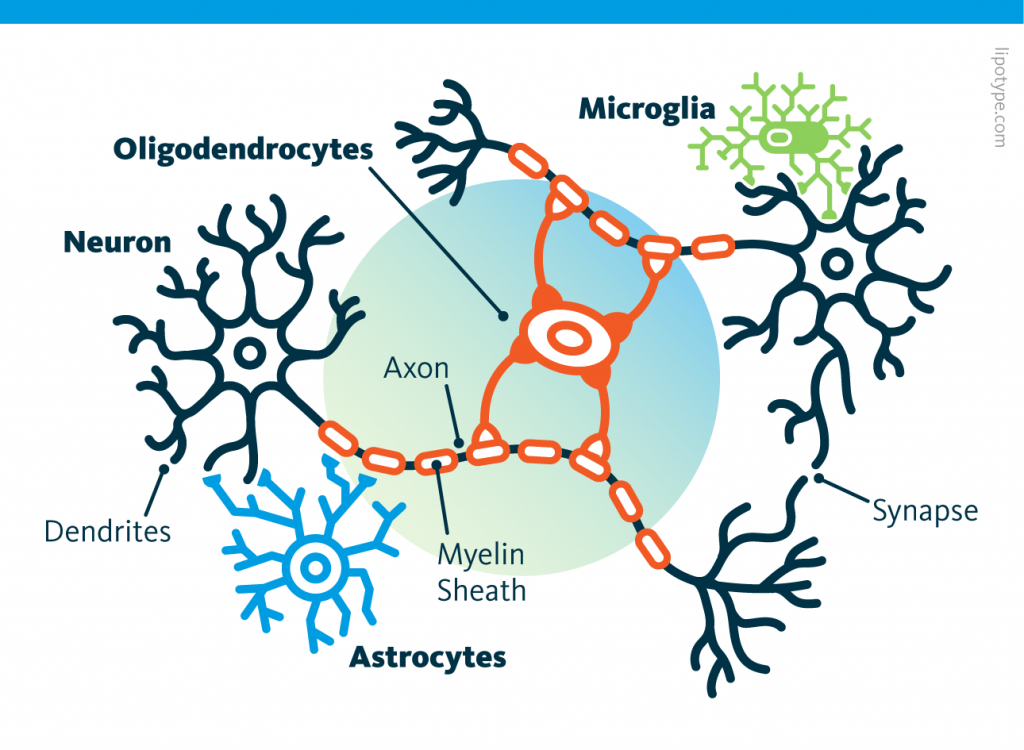 Curiously, scientists are rather looking to directly convert astrocytes into neurons, despite the enormous morphological difference between these two types of cells.
Curiously, scientists are rather looking to directly convert astrocytes into neurons, despite the enormous morphological difference between these two types of cells.