Peut-on « restaurer » le cerveau atteint d’Alzheimer au moyen de NAD+ ?
La maladie d’Alzheimer (MA) a longtemps été considérée comme le « cimetière de la découverte de médicaments ». Pendant des décennies, les efforts se sont concentrés sur l’élimination des plaques amyloïdes, véritables « déchets » du cerveau. Il y a eu plus de 1500 essais cliniques sur la maladie d’Alzheimer. Cela représente des investissements colossaux en temps et en argent.
Il se dit que les scientifiques des entreprises du secteur recevoient une prime s'ils obtiennent un médicament candidat ayant franchi l'étape des études animales pour atteindre les essais cliniques chez l'humain. Un moyen d'obtenir « systématiquement » cette prime est de travailler sur des médicaments candidats pour les maladies neurologiques (par exemple, la maladie d'Alzheimer) car les modèles animaux pour ces maladies « guérissent » la maladie neurologique étudiée chez la souris, le rat, etc. Cela permet aussi d'induire en erreur les éventuels investisseurs.
Mais une étude récente et médiatisée sur un composé appelé P7C3-A20 déplace le débat : on passe du « nettoyage des déchets » à la « réparation du cœur même de la cellule ». https://www.cell.com/cell-reports-medicine/fulltext/S2666-3791(25)00608-1
Voici un aperçu de cette recherche des principes scientifiques qui la sous-tendent et des raisons pour lesquelles il convient de rester prudent.
La recherche met en évidence une baisse critique du NAD+ (nicotinamide adénine dinucléotide), une molécule indispensable à la production d’énergie et à la réparation cellulaire. Chez les patients atteints de MA et chez les souris « 5xFAD », les niveaux de NAD+ chuteraient de près de 45 % à mesure que la maladie progresse. Il ne s'agit que d'une simple corrélation qui est vrai pour des centaines d'autres molécules.
L'étude a testé le P7C3-A20, une molécule conçue pour normaliser les niveaux de NAD+. Les résultats obtenus chez les souris étaient tout apparement simplement « miraculeux ».
Inversion des pertes de mémoire : des souris présentant un déclin cognitif avancé auraient retrouvé la capacité d'apprendre et de se déplacer.
Réparation de la barrière hémato-encéphalique : la molécule aurait réparé la perméabilité de la barrière hémato-encéphalique (BHE), empêchant ainsi l'infiltration de toxines.
Atténuation de l'inflammation : les cellules immunitaires « toxiques » du cerveau auraient retrouvé un état protecteur.
Restauration de l'énergie : en réparant les mitochondries (les centrales énergétiques de la cellule), le médicament aurait permis au cerveau d'éliminer plus efficacement ses déchets amyloïdes et tau.
Le problème des « molécules miracles » : Est-ce trop beau pour être vrai ?
Lorsqu'un médicament agit simultanément sur cinq systèmes biologiques sans lien apparent — inflammation, mémoire, barrière hémato-encéphalique, dommages à l'ADN et repliement des protéines —, les scientifiques (et les patients) devraient se poser des questions essentielles.
- Le signal d’alarme du « pitch deck »
L’étude est remarquablement bien présentée, affichant tous les « succès » qu’un investisseur en biotechnologies pourrait souhaiter. Étant donné que certains des auteurs ont des liens avec les entreprises développant ces molécules, il existe un risque de « biais de publication », où seuls les résultats les plus parfaits sont rendus publics.
- Les souris ne sont pas des humains
Nous avons guéri la maladie d’Alzheimer chez la souris des centaines de fois. Cependant, les souris génétiquement modifiées développent une maladie en quelques mois qui a des poimts communs avec la maladie d’Alzheimer humaine mais posséde 1000 fois moins de neurones qu'un humaim. La maladie d’Alzheimer chez l’humain est un processus complexe qui s’étend sur 20 ans et qui implique le vieillissement, le mode de vie et la dégénérescence vasculaire. Ce qui fonctionne chez une souris de 12 mois échoue le plus souvent (toujours) chez un humain de 80 ans.
- Inversion ou régénération
Si la maladie d’Alzheimer détruit les neurones, comment peut-on l’« inverser » ? Cette publication ne prétend pas régénérer le cerveau. Elle suggère que de nombreux neurones « malades » ne sont pas morts ; qu'ils seraient simplement « mis en veille » par manque de NAD+ et que le P7C3-A20 réactiverait efficacement le réseau neuronal survivant. Cela n'est guére crédible, un cerveau de malade présente des zones clairement détruites.
Comparaison avec le NR et le NMN :
Vous avez peut-être entendu parler de suppléments stimulant le NAD+ comme le nicotinamide riboside (NR) ou le NMN. Bien que populaires, leur efficacité lors des essais cliniques chez l’humain pour la maladie d’Alzheimer n'a pas été prouvée.
Le NR et le NMN fournissent les « matières premières » (les briques). Si le fonctionnement du cerveau est altéré, il ne peut pas les utiliser. Les administrer à un cerveau malade revient à livrer des briques à un chantier où les ouvriers sont en grève. Le P7C3-A20 est différent : c’est un « activateur » qui cible les enzymes (les ouvriers) pour qu’elles utilisent plus efficacement le carburant disponible.
Conclusion
Les travaux sur le P7C3-A20 offrent une perspective fascinante, identifiant 17 protéines spécifiques « défectueuses » chez la souris et chez l’humain. Bien que cela puisse ressembler à une présentation commerciale pour un nouveau médicament, l'approche axée sur la résilience métabolique – rendre le cerveau plus résistant plutôt que simplement plus propre – représente un tournant intéressant dans la lutte contre la maladie d'Alzheimer.
En attendant les résultats d'un essai clinique de phase II chez l'humain, il nous faut nuancer notre espoir face à la réalité : le cerveau humain est une structure extremement complexe, et optimiser son fonctionnement est une tâche ardue.

 By Manu5 - http://www.scientificanimations.com/wiki-images/
By Manu5 - http://www.scientificanimations.com/wiki-images/ Ils ont confirmé qu'une exposition prolongée aux NRTI était associée à un risque moindre de développer la maladie d’Alzheimer.
- D’après les données du VA, chaque année supplémentaire de traitement par NRTI était associée à une réduction de 4 à 6 % du risque de maladie d’Alzheimer.
- D’après les données de MarketScan, la réduction était encore plus marquée : de 10 à 13 % par année d’utilisation.
Ils ont confirmé qu'une exposition prolongée aux NRTI était associée à un risque moindre de développer la maladie d’Alzheimer.
- D’après les données du VA, chaque année supplémentaire de traitement par NRTI était associée à une réduction de 4 à 6 % du risque de maladie d’Alzheimer.
- D’après les données de MarketScan, la réduction était encore plus marquée : de 10 à 13 % par année d’utilisation.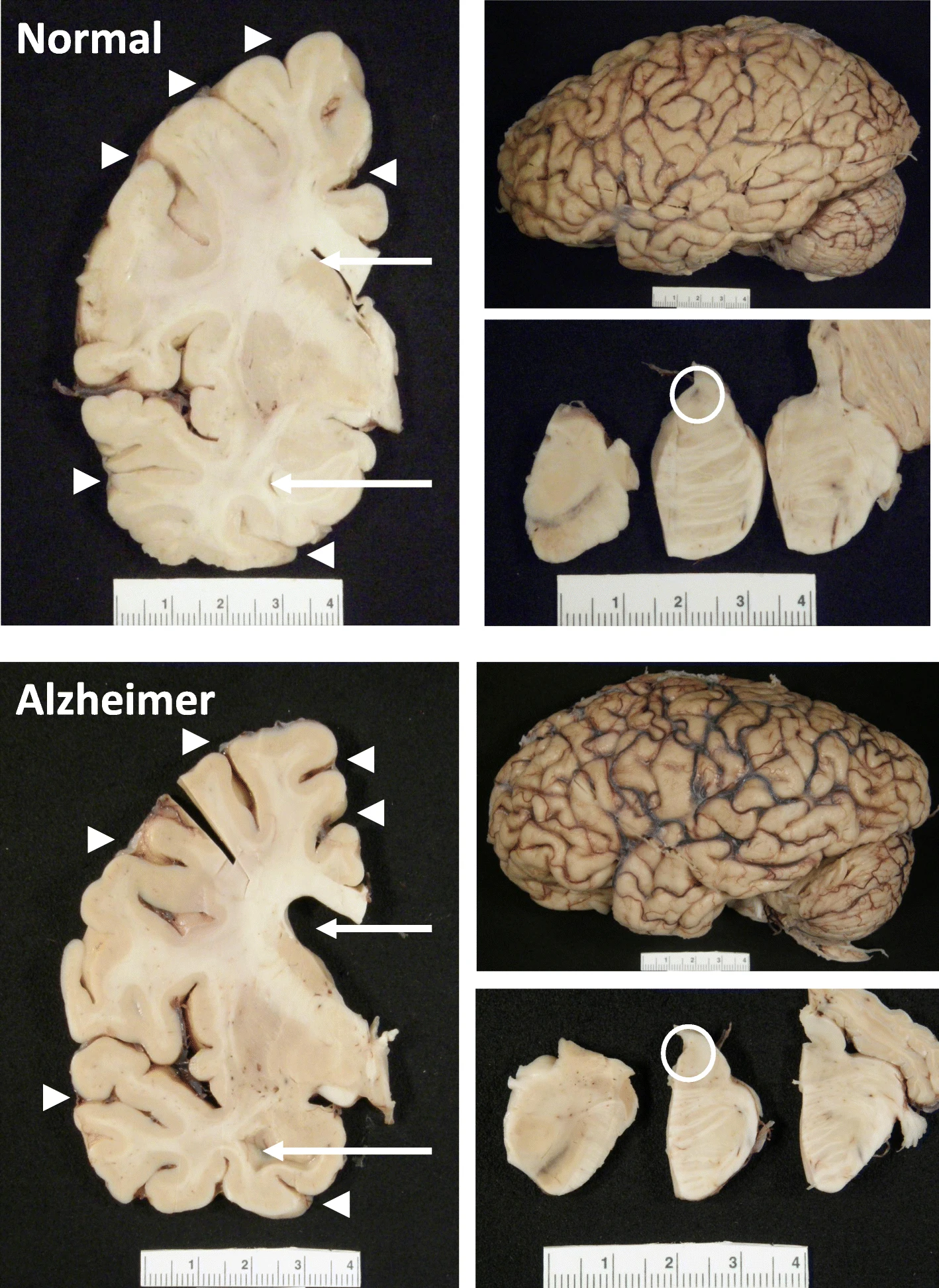 The hippocampus, a small, seahorse-shaped structure buried deep within the brain, is best known for its role in memory formation and learning. It is an exceptionally vulnerable structure, with perfusion deficits often observed in diseases related to learning and memory. However, a brain affected by Alzheimer's disease tends to exhibit at least moderate cortical atrophy, including in the precuneus and posterior cingulate gyrus. It should be noted that the posterior cingulate gyrus is adjacent to the hippocampus.
The hippocampus, a small, seahorse-shaped structure buried deep within the brain, is best known for its role in memory formation and learning. It is an exceptionally vulnerable structure, with perfusion deficits often observed in diseases related to learning and memory. However, a brain affected by Alzheimer's disease tends to exhibit at least moderate cortical atrophy, including in the precuneus and posterior cingulate gyrus. It should be noted that the posterior cingulate gyrus is adjacent to the hippocampus.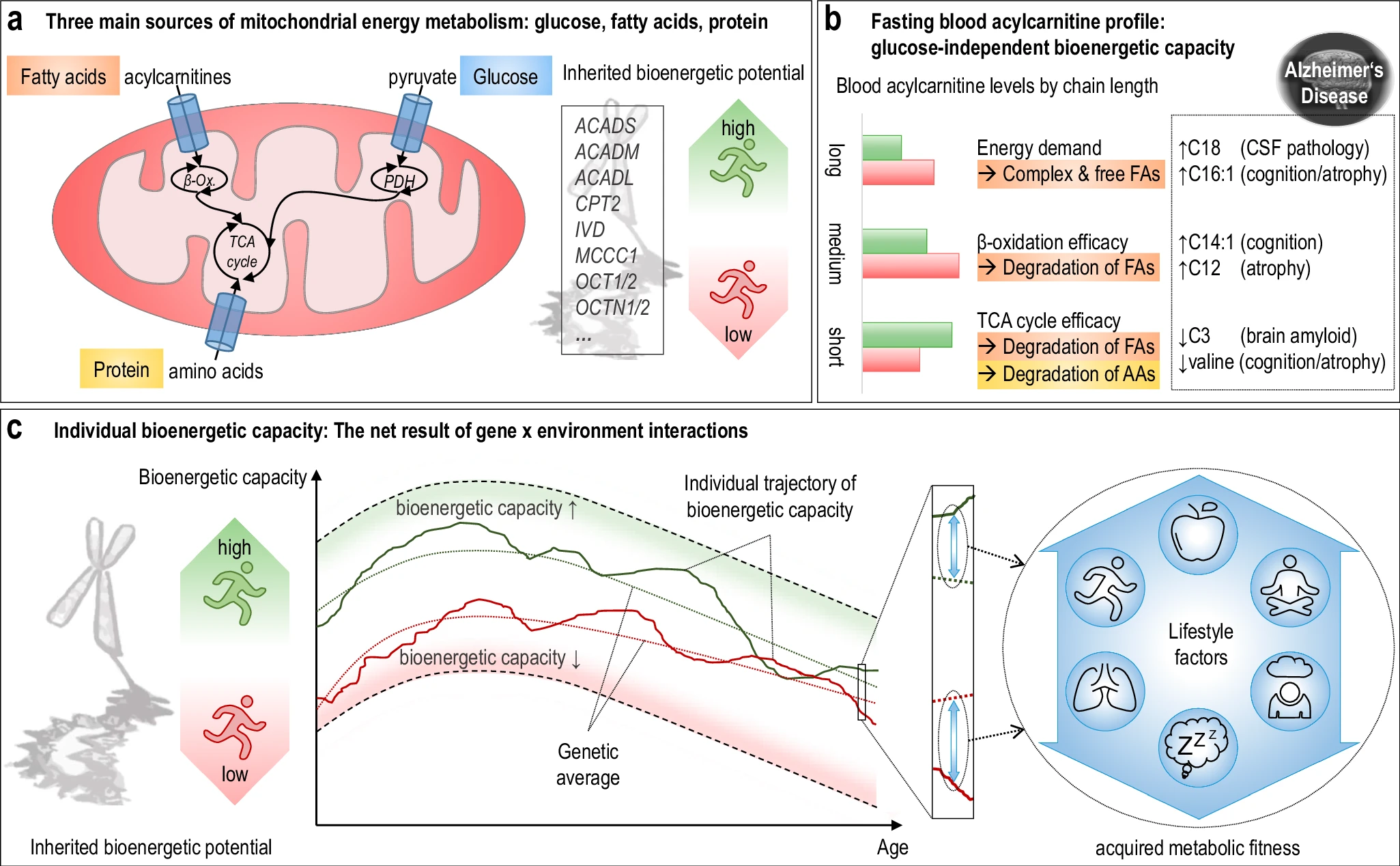 The researchers used acylcarnitine profiles from blood samples to identify distinct bioenergetic subgroups in Alzheimer's Disease (AD) patients and evaluate how bioenergetic capacity relates to disease progression.
They used data from 1,531 participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI), and identified several bioenergetic subgroups with significant differences in AD biomarkers, cognitive function, and brain glucose metabolism.
These subgroups were primarily determined by modifiable factors (40-60%) related to beta-oxidation function, rather than genetic factors, suggesting potential for intervention.
The researchers used acylcarnitine profiles from blood samples to identify distinct bioenergetic subgroups in Alzheimer's Disease (AD) patients and evaluate how bioenergetic capacity relates to disease progression.
They used data from 1,531 participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI), and identified several bioenergetic subgroups with significant differences in AD biomarkers, cognitive function, and brain glucose metabolism.
These subgroups were primarily determined by modifiable factors (40-60%) related to beta-oxidation function, rather than genetic factors, suggesting potential for intervention.