Recent research continues to highlight the complex relationship between cardiovascular health and brain aging. For instance, vascular dementia is one type of dementia. Furthermore, universities are encouraging their teams to identify useful biomarkers for neurodegenerative diseases, as they believe they can generate revenue from potential patents. One quick and inexpensive method to conduct a study is to utilize databases available online.
The Framingham Study is a long-term epidemiological study (since 1948) that initially focused on cardiovascular disease. We owe much of our knowledge about cardiovascular disease to the Framingham Study, particularly regarding the identification of cardiovascular risk factors, including the effects of smoking and diet. Thus, the recognition of high blood pressure as a risk factor for heart disease dates back to 1957, for neurological diseases to 1967, and heart failure to 1971.
 By Manu5 - http://www.scientificanimations.com/wiki-images/
By Manu5 - http://www.scientificanimations.com/wiki-images/
At the same time, the Framingham Study focused on a relatively affluent population of European origin, meaning the conclusions drawn from it may not necessarily apply to other populations. Furthermore, marker values vary depending on the data collection devices and methods (some values date back to 1948!), so an a priori analysis must consider these difficulties, which is rarely addressed in rapid studies.
The authors of the article discussed in this post selected 822 elderly individuals (mean age: approximately 72 years) from the database who were not suffering from dementia at the time of their first blood test with the Framingham Study. Participants were monitored for Alzheimer's disease until 2020. Over a median follow-up period of 12.5 years, 128 of these individuals developed Alzheimer's disease.
The article authors measured levels of high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), small dense LDL cholesterol (sdLDL-C), lipoprotein a (Lp(a)), apolipoprotein B (ApoB), and the ApoB48 isoform of ApoB in the blood samples collected from 1985 to 1988 by the Framingham Study.
It has long been acknowledged that, although the mechanism is not fully understood, elevated levels of apolipoprotein B (ApoB) are associated with higher concentrations of LDL particles and are the primary driver of plaques that cause vascular disease (atherosclerosis), which typically first manifests as obvious symptoms such as heart disease, stroke, and various other body-wide complications after decades of progression.
Furthermore, the causes of Alzheimer's disease remain poorly understood, but the most significant genetic risk factor arises from an allele of apolipoprotein E. Other risk factors include a history of head trauma, clinical depression, and high blood pressure. The progression of the disease is largely characterized by the accumulation of malformed protein deposits in the cerebral cortex, known as amyloid plaques and neurofibrillary tangles.
Therefore, there is a possible link between dementia and cardiovascular risk, and the Framingham Study database allows us to statistically compare heart health and the risk of developing comorbidities.
The article authors considered a wide range of cholesterol-related markers:
HDL-C: "Good" cholesterol
LDL-C: "Bad" cholesterol
sdLDL-C: Small, dense particles of LDL cholesterol
ApoB and ApoB48: Proteins involved in cholesterol transport
Lp(a): A lipoprotein linked to the risk of heart disease
The researchers examined the relationship between these markers, standardized by their standard deviation units (SDUs), and the future risk of Alzheimer's disease.
** sdLDL-C ** They found that high sdLDL-C levels correlate with an increased risk of developing Alzheimer's disease. This result has also been observed by other teams, including a recent one that examined a database in Finland:
SdLDL-C stands for "small dense low-density lipoprotein cholesterol." It is a subclass of LDL particles that are smaller and denser than conventional LDL and more likely to penetrate blood vessel walls. This cholesterol subtype is rarely tested. sdLDL is regarded as a more atherogenic lipoprotein due to its higher retention rate in the arterial wall, better penetration, lower binding affinity to the LDL receptor, lower resistance to oxidative stress, and longer plasma half-life. In addition to being associated with a heightened risk of cardiovascular disease, sdLDL, particularly sdLDL-C levels, have been linked to type 2 diabetes mellitus, metabolic syndrome, obesity, and low-grade inflammation.
** ApoB48 ** Conversely, elevated ApoB48 levels are linked to a reduced risk of Alzheimer's disease. This finding was previously known.
** HDL-C ** However, the authors uncovered surprising results regarding HDL-C, often referred to as "good cholesterol". Numerous epidemiological studies have indicated that high circulating HDL levels are associated with a reduced risk of Alzheimer's disease. Yet, this new study's authors found that participants with the lowest HDL-C levels had a 44% reduced risk of developing Alzheimer's disease compared to those with higher HDL-C levels.
Because HDL-C is generally considered protective, this team's findings may reflect other age-related changes, such as metabolic status or weight loss, that occur before the onset of dementia.
 Ils ont confirmé qu'une exposition prolongée aux NRTI était associée à un risque moindre de développer la maladie d’Alzheimer.
- D’après les données du VA, chaque année supplémentaire de traitement par NRTI était associée à une réduction de 4 à 6 % du risque de maladie d’Alzheimer.
- D’après les données de MarketScan, la réduction était encore plus marquée : de 10 à 13 % par année d’utilisation.
Ils ont confirmé qu'une exposition prolongée aux NRTI était associée à un risque moindre de développer la maladie d’Alzheimer.
- D’après les données du VA, chaque année supplémentaire de traitement par NRTI était associée à une réduction de 4 à 6 % du risque de maladie d’Alzheimer.
- D’après les données de MarketScan, la réduction était encore plus marquée : de 10 à 13 % par année d’utilisation.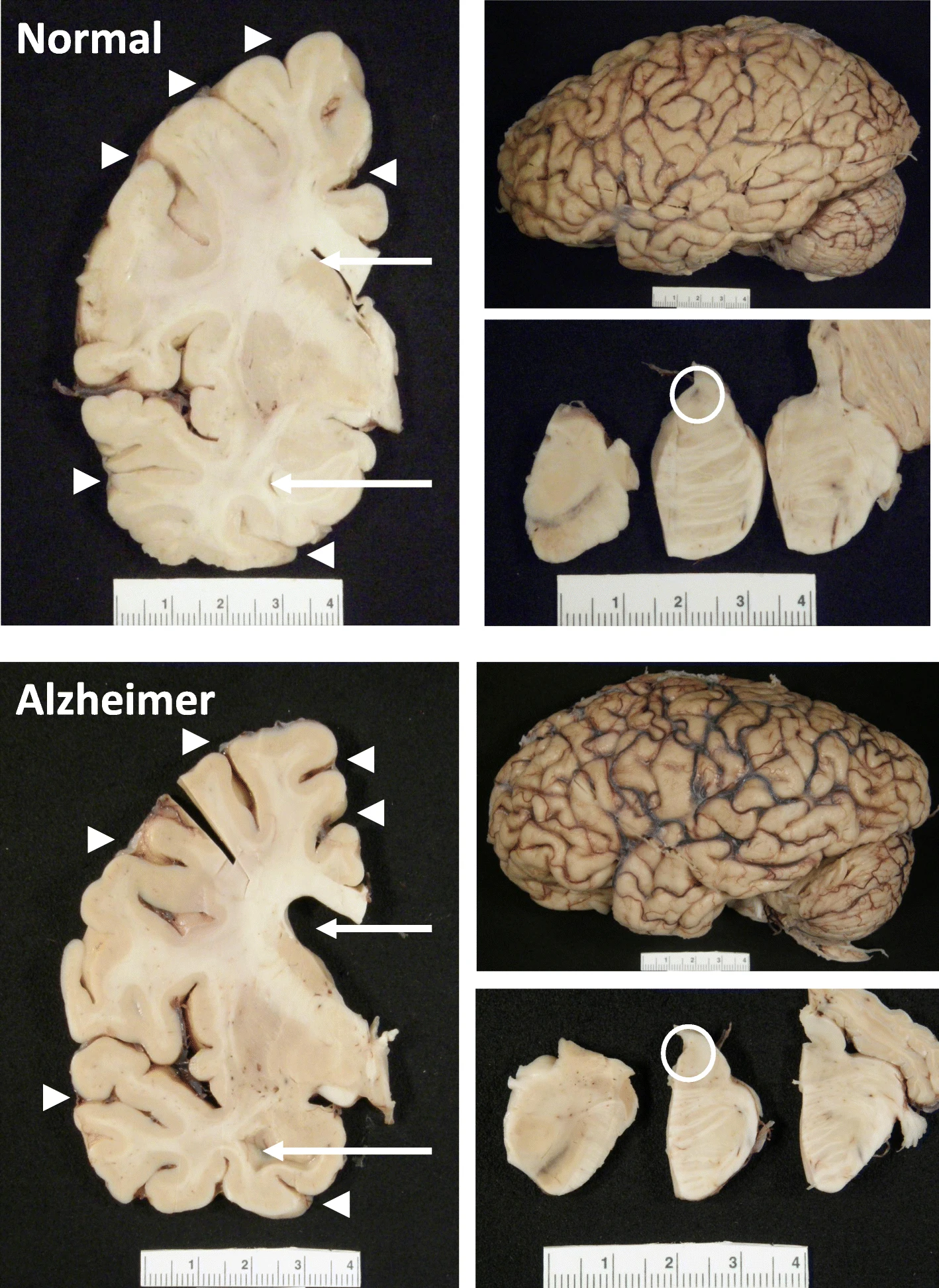 The hippocampus, a small, seahorse-shaped structure buried deep within the brain, is best known for its role in memory formation and learning. It is an exceptionally vulnerable structure, with perfusion deficits often observed in diseases related to learning and memory. However, a brain affected by Alzheimer's disease tends to exhibit at least moderate cortical atrophy, including in the precuneus and posterior cingulate gyrus. It should be noted that the posterior cingulate gyrus is adjacent to the hippocampus.
The hippocampus, a small, seahorse-shaped structure buried deep within the brain, is best known for its role in memory formation and learning. It is an exceptionally vulnerable structure, with perfusion deficits often observed in diseases related to learning and memory. However, a brain affected by Alzheimer's disease tends to exhibit at least moderate cortical atrophy, including in the precuneus and posterior cingulate gyrus. It should be noted that the posterior cingulate gyrus is adjacent to the hippocampus.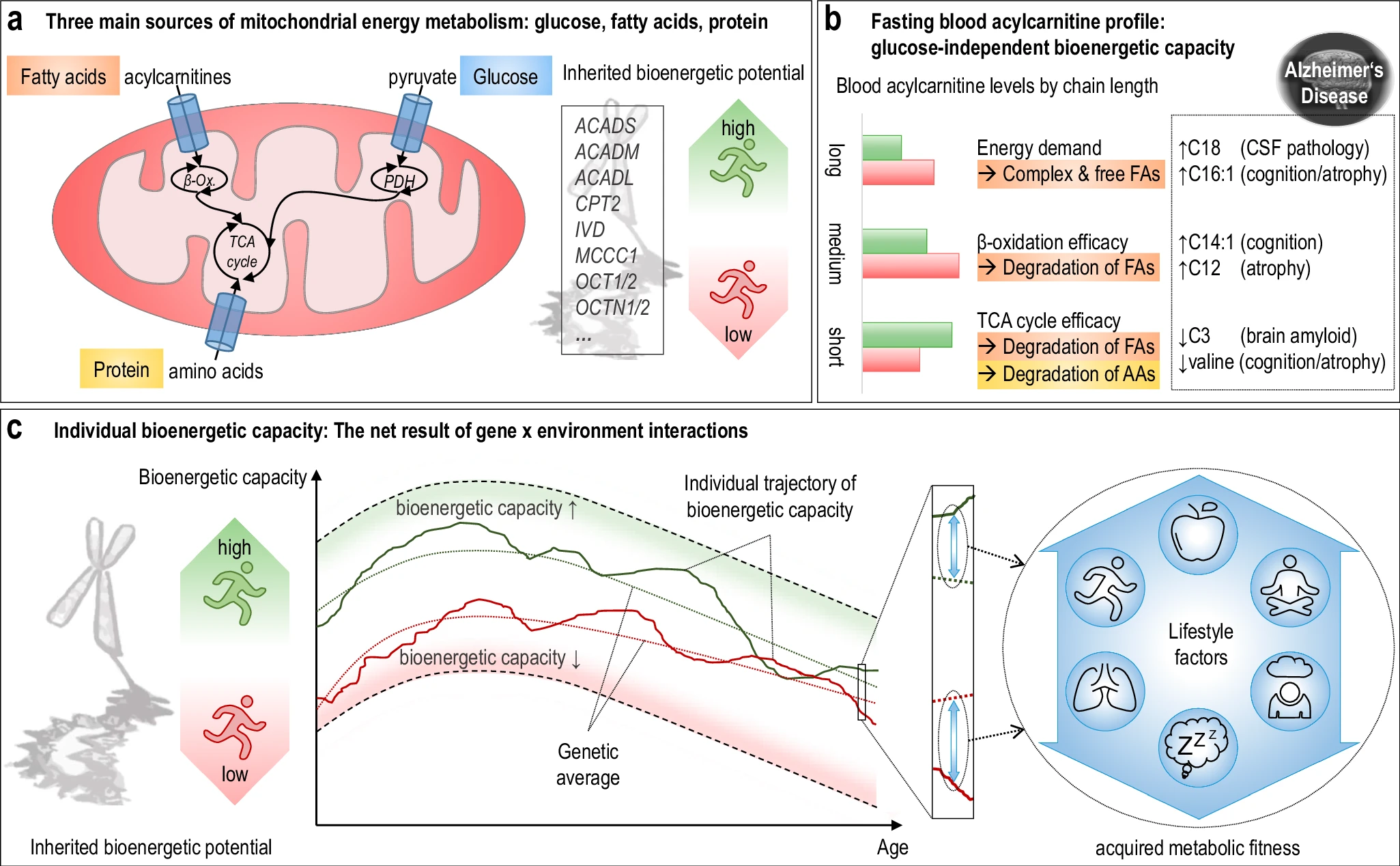 The researchers used acylcarnitine profiles from blood samples to identify distinct bioenergetic subgroups in Alzheimer's Disease (AD) patients and evaluate how bioenergetic capacity relates to disease progression.
They used data from 1,531 participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI), and identified several bioenergetic subgroups with significant differences in AD biomarkers, cognitive function, and brain glucose metabolism.
These subgroups were primarily determined by modifiable factors (40-60%) related to beta-oxidation function, rather than genetic factors, suggesting potential for intervention.
The researchers used acylcarnitine profiles from blood samples to identify distinct bioenergetic subgroups in Alzheimer's Disease (AD) patients and evaluate how bioenergetic capacity relates to disease progression.
They used data from 1,531 participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI), and identified several bioenergetic subgroups with significant differences in AD biomarkers, cognitive function, and brain glucose metabolism.
These subgroups were primarily determined by modifiable factors (40-60%) related to beta-oxidation function, rather than genetic factors, suggesting potential for intervention. Alzheimer's disease research has produced many hypotheses over the years, including cholinergic, inflammatory, viral, mitochondrial, tau, and amyloid. However, none of these hypotheses have led to treatments that can stop or reverse the disease. This leads to a search for new theories to explain these failures. But this may be because interventions occur too late in the disease progression, with brain damage irreparable and compensatory mechanisms saturated.
Alzheimer's disease research has produced many hypotheses over the years, including cholinergic, inflammatory, viral, mitochondrial, tau, and amyloid. However, none of these hypotheses have led to treatments that can stop or reverse the disease. This leads to a search for new theories to explain these failures. But this may be because interventions occur too late in the disease progression, with brain damage irreparable and compensatory mechanisms saturated.
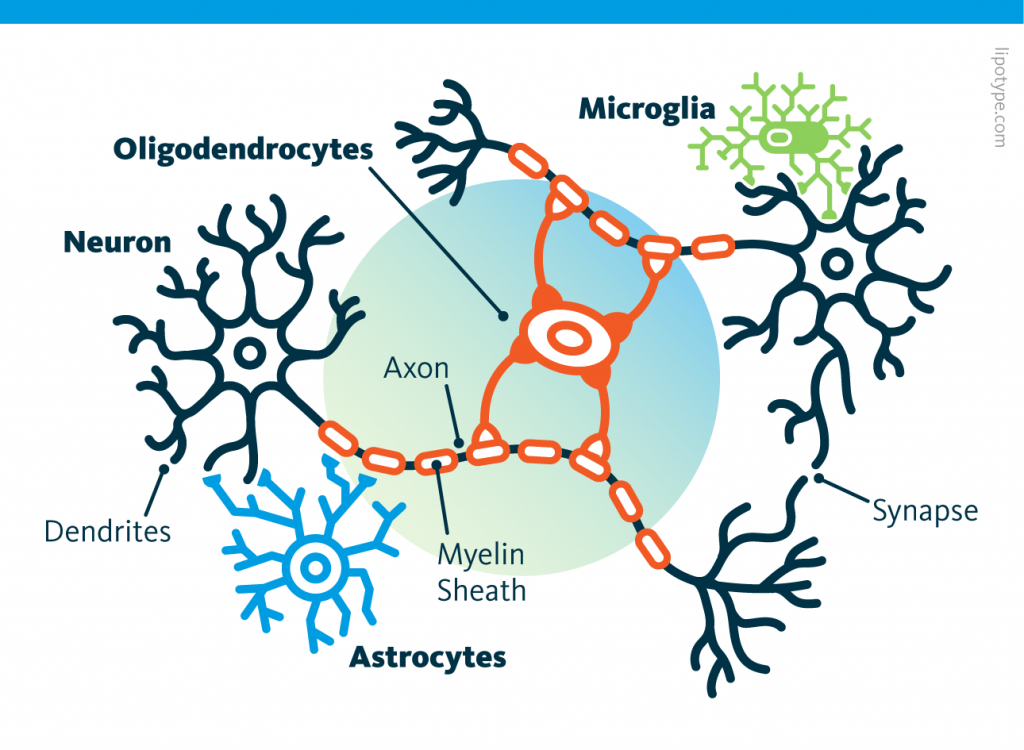 Curiously, scientists are rather looking to directly convert astrocytes into neurons, despite the enormous morphological difference between these two types of cells.
Curiously, scientists are rather looking to directly convert astrocytes into neurons, despite the enormous morphological difference between these two types of cells. Those other cells, which compose half of the brain's cells, are receiving more attention. There are multiple types but normally they are there to assist neurons in their task. A simplified view tells that neurons are a sort of plumbing system and the glial cells are the real actors in the brain.
Those other cells, which compose half of the brain's cells, are receiving more attention. There are multiple types but normally they are there to assist neurons in their task. A simplified view tells that neurons are a sort of plumbing system and the glial cells are the real actors in the brain. Source: Nephron via Wikipedia
Source: Nephron via Wikipedia
 Source: Peta
Source: Peta