L'invasion placentaire dans l'endomètre maternel de l'utérus présente des similitudes substantielles avec la dissémination précoce du cancer dans le stroma (la partie d'un tissu ou d'un organe ayant un rôle structurel ou conjonctif).
Ces similitudes ont inspiré l'hypothèse que les trophoblastes (la couche cellulaire continue formée de fibroblastes qui limite l'œuf, devenu blastocyste au 6e jour après la fécondation) ont développé la capacité d'envahir l'endomètre, conduisant à une placentation invasive. L'invasion d'un type spécifique de trophoblaste (trophoblaste extravilleux) dans l'utérus maternel est une étape vitale dans l'établissement de la grossesse.
 Source Zephyris CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=10811330
Source Zephyris CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=10811330
Ces mécanismes peuvent se réactiver dans les cellules cancéreuses, entraînant une prédisposition aux métastases. Il avait donc été fait l'hypothèse (nommée ELI) que la malignité cancéreuse devait être limitée aux mammifères placentaires où la placentation invasive a d'abord évolué. Mais il y a plusieurs contre-exemples.
Dans un article récent, les auteurs explorent un scénario alternatif dans lequel les cellules stromales de l'utérus ont évolué pour résister ou permettre l'invasion, déterminant le résultat de l'invasivité placentaire. La probabilité que l'évolution de l'environnement stromal entraîne l'évolution de la malignité cancéreuse est renforcée par le fait que les mécanismes moléculaires utilisés par les cellules cancéreuses pour métastaser sont partagés avec d'autres processus biologiques.
Par exemple, les mécanismes régulant la gastrulation, la cicatrisation des plaies, l'extravasation par les leucocytes, etc., sont partagés à la fois avec le trophoblaste et l'invasion du cancer. Cela implique que les cellules cancéreuses envahissantes utilisent des mécanismes qui ont évolué beaucoup plus tôt que l'invasion placentaire et, par conséquent, l'évolution de la placentation invasive en soi ne peut pas être responsable de l'origine du cancer malin.
Il est important de noter, cependant, que la nature invasive du placenta a continué d'évoluer après son origine. Les espèces de mammifères diffèrent par leur potentiel de tumorigenèse, ainsi que leur vulnérabilité aux métastases cancéreuses.
Alors que l'évolution a entraîné un degré d'envahissement encore plus élevé chez les grands singes, qui comprend les humains, une perte complète de l'invasion placentaire a évolué chez les mammifères à sabots, tels que les vaches et les chevaux et leurs parents, et ces animaux ont des taux de malignité inférieurs pour une variété des cancers.
Dans une revue récente, Constanzo et ses collaborateurs ont présenté des arguments convaincants pour un modèle où la progression du cancer chez l'homme comprend la réactivation de l'expression des gènes embryonnaires contrôlant normalement le développement du placenta et l'évasion immunitaire.
Par exemple, le mélanome survient chez les bovins et les équidés mais reste largement bénin; alors qu'il est très malin chez l'homme. Ceci est en corrélation avec le phénotype de l'interface fœtale-maternelle (le degré d'invasion placentaire pendant la grossesse).
En particulier, ces résultats soutiennent que la sécrétion de TGF-β et la signalisation WNT non canonique élevée dans les cellules stromales sont des facteurs causaux expliquant la forte vulnérabilité des tissus stromaux humains à l'invasion du cancer, au moins dans le cas du mélanome.
Leurs données soutiennent l'hypothèse ELI, suggérant que les différences d'expression génique stromale entre les espèces sont déterminantes pour déterminer le degré d'implantation de l'embryon ainsi que la résistance stromale à la dissémination précoce du cancer.

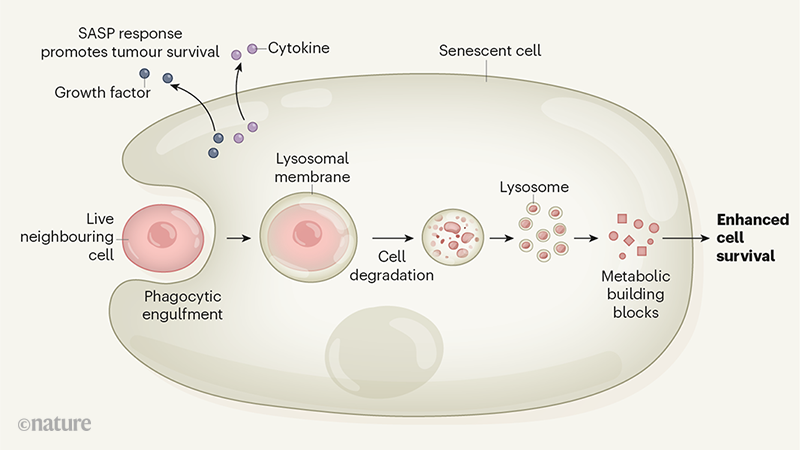

 * By BrainsRusDC - Personal work, CC BY 4.0, https://commons.wikimedia.org/w/index.php?curid=64271015*
* By BrainsRusDC - Personal work, CC BY 4.0, https://commons.wikimedia.org/w/index.php?curid=64271015* Source: Blausen.com staff (2014). "Medical gallery
Source: Blausen.com staff (2014). "Medical gallery