Mitochondria, which are responsible for producing energy in cells, play a crucial role in neuronal function. Neurons have high energy requirements, and mitochondrial dysfunction can contribute to the selective degeneration of motor neurons in ALS. In ALS, there is evidence of altered mitochondrial metabolism and impaired mitochondrial transport within neurons.
 The primary function of mitochondria in normal cells is to employ oxidative phosphorylation of ADP via the electron transport chain to produce ATP. Compared to non-excitable cells such as fibroblasts (a standard experimental platform for studying mitochondrial dynamics), neurons are especially dependent upon mitochondrial ATP because of the high energy requirements for electrochemical neurotransmission.
The primary function of mitochondria in normal cells is to employ oxidative phosphorylation of ADP via the electron transport chain to produce ATP. Compared to non-excitable cells such as fibroblasts (a standard experimental platform for studying mitochondrial dynamics), neurons are especially dependent upon mitochondrial ATP because of the high energy requirements for electrochemical neurotransmission.
In ALS, the requirement for constant ATP production may be compounded by altered or impaired mitochondrial metabolism, as reported in pre-clinical models. The unique elongated anatomy of upper and lower motor neurons poses special challenges for ATP delivery, which may contribute to the motor neuron selectivity of ALS.
Because ATP undergoes spontaneous hydrolysis in an aqueous solution at physiological pH, mitochondria need to be positioned at subcellular locations proximate to ATP utilization.
While the generation of “new” mitochondria to replace those lost to damage or senescence necessarily occurs in neuronal soma, mitochondrial residence within the distal synaptic branches of normal neurons (where ATP production fuels neuronal signaling) and in the terminal growth buds of damaged/regenerating axons (where ATP production fuels neuronal repair and regrowth) is essential for neuronal homeostasis.. Thus, the mitochondrial supply chain is long and fragile.
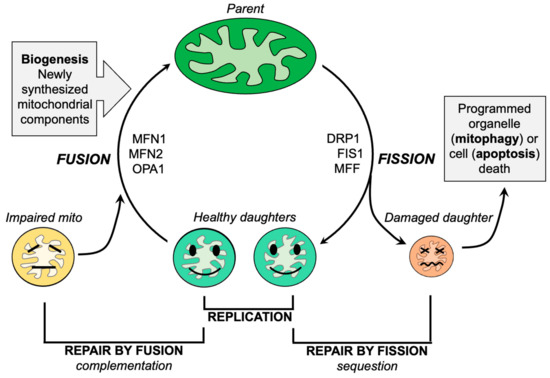
Mitochondrial dysdynamism refers to the imbalance between mitochondrial fusion (the joining of mitochondria) and fission (the division of mitochondria) processes. In ALS, there is a decrease in fusion-promoting proteins and an increase in fission-related proteins, suggesting an imbalance in mitochondrial dynamics. Additionally, impaired mitochondrial motility is observed in ALS, which further affects their proper distribution within neurons.
Like everything, mitochondria wear out. Because >99% of their protein components are transcribed in the nucleus and translated in neuronal soma, the functional lifespan of a mitochondrion located at a distal synaptic terminus can be abbreviated. Moreover, as neurons are terminally differentiated cells, mitochondrial renewal during cell proliferation does not occur.
Healthy mitochondria face a formidable physical challenges in the anterograde transport of young mitochondria, from the soma to distal motor neuron synapses which must be balanced by removing damaged, potentially cytotoxic mitochondria from distal neuronal termini through retrograde transport. It is not surprising that mitochondrial dysmotility and clumping within neuronal soma (like traffic congestion, a sign of impaired transport) are observed in many neurodegenerative diseases, including ALS
In conclusion, one approach is to inhibit mitochondrial fission using small molecules or peptides, which has shown promise in reversing mitochondrial fragmentation and improving motor function in animal models of ALS. Another approach is to enhance mitochondrial fusion by activating mitofusin proteins, which has also demonstrated positive effects in improving mitochondrial morphology and delaying disease progression in animal models.