Voici un article récent qui fournit des informations intéressantes sur une caractéristique commune à plus de la moitié des des malades de la SLA: L'hypermétabolisme.
Il semble paradoxal que les malades de la SLA qui perdent progressivement leurs muscles soient atteint d'hypermétabolisme. Cet article, qui n'est en rien à propos des maladies neurodégénératives, explique celà et plus encore, il fait le lien avec le stress cellulaire qui induit des granules de protéines déformées et mal localisées. Cet article me plaît aussi car il n'assume pas que les processus biologiques n'existent ex-nihilo (au contraire de la quasi-totalité des publications) mais nécessitent de l'énergie et sont en concurrence les uns avec les autres. Enfin le rôle néfaste de l'alcool y est abordé.
 Dans la biochimie de tous les êtres vivants connus, l'ATP fournit l'énergie nécessaire aux réactions chimiques du métabolisme. L'essentiel de l'ATP produit par les eucaryotes non photosynthétiques provient de la phosphorylation oxydative au sein des mitochondries.
Dans la biochimie de tous les êtres vivants connus, l'ATP fournit l'énergie nécessaire aux réactions chimiques du métabolisme. L'essentiel de l'ATP produit par les eucaryotes non photosynthétiques provient de la phosphorylation oxydative au sein des mitochondries.
Les maladies mitochondriales sont causées par des mutations dans le génome mitochondrial ou cellulaire, qui altèrent la phosphorylation oxydative (OxPhos) et la capacité de convertir les substrats alimentaires en ATP. Dans les modèles animaux, les défauts OxPhos déclenchent des réponses transcriptionnelles nucléaires, y compris la réponse intégrée au stress (ISR). Ces voies de réponse au stress impliquent des processus cellulaires fondamentalement exigeants en énergie et augmente la consommation d'énergie au niveau cellulaire et de l'organisme.
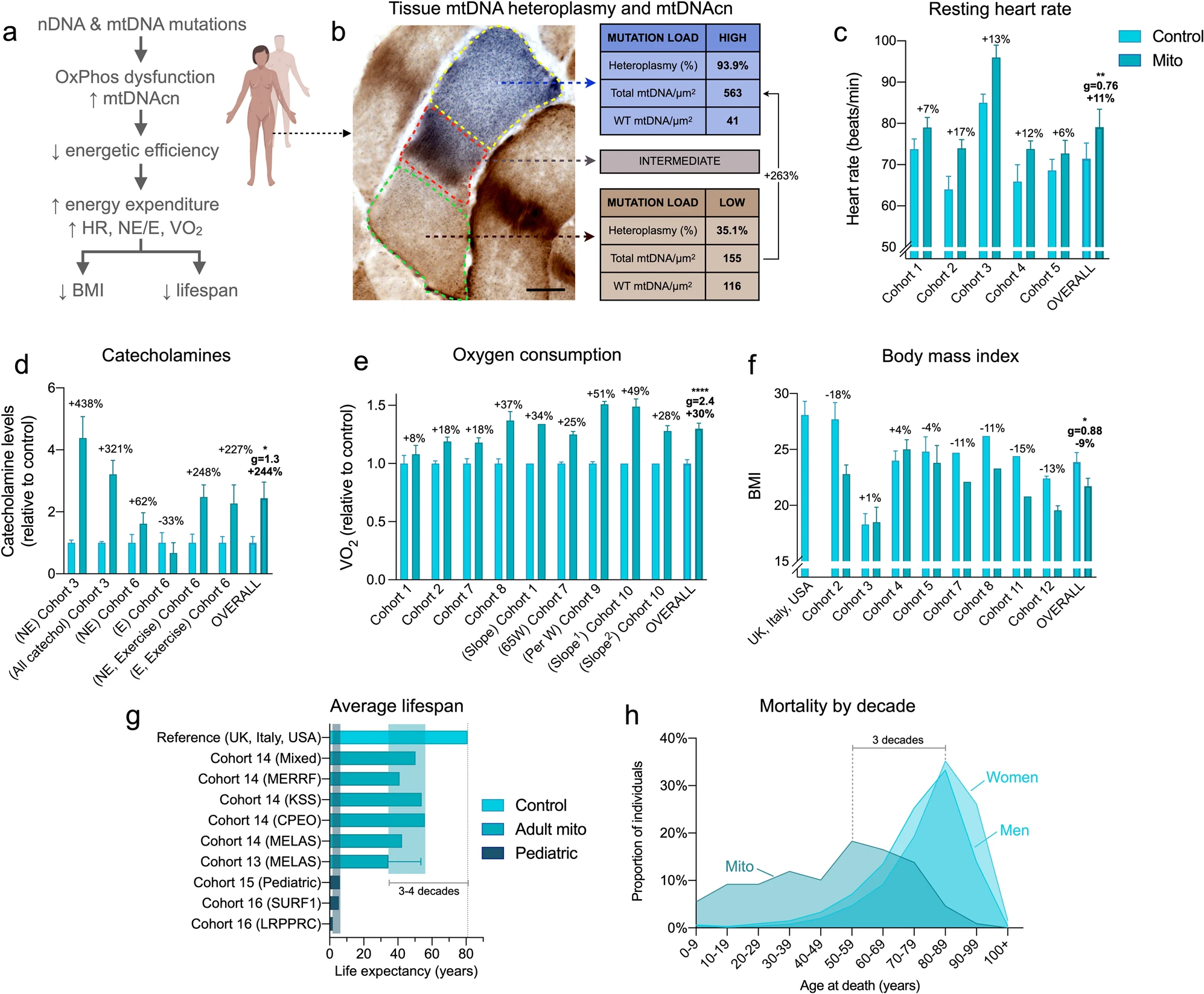
Une idée fausse courante est qu'une capacité mitochondriale réduite à oxyder les substrats associée à des niveaux d'activité physique minimaux favoriserait une réponse de conservation de l'énergie, entraînant un bilan énergétique positif et une accumulation de graisse corporelle, conduisant à l'obésité. Cependant, les patients atteints de maladies mitochondriales sont rarement obèses. En fait, les patients atteints d'une maladie modérée à sévère sont en moyenne classés comme ayant un poids insuffisant.
La rareté de l'obésité dans les maladies mitochondriales reste un paradoxe clinique. Cela peut être résolu par la notion contre-intuitive selon laquelle les défauts mitochondriaux d'OxPhos peuvent ne pas diminuer la consommation et les dépenses d'énergie, mais au contraire peuvent augmenter le coût énergétique nécessaire pour maintenir les fonctions physiologiques de base.
Au sein des cellules, les coûts métaboliques proviennent principalement des processus de transcription/traduction (~60 % des besoins énergétiques totaux), du maintien de l'équilibre ionique, ainsi que de la biogenèse et de la dégradation des organelles, qui comprend le renouvellement mitochondrial. Par conséquent, la biogenèse mitochondriale entraîne un coût énergétique important en raison de l'important protéome mitochondrial19.
Une analyse de la dépense énergétique au repos (REE) dans des modèles animaux présentant des défauts OxPhos mitochondriaux indique que la dépense énergétique au repos est probablement élevée de 15 à 85 %.
Par conséquent, les auteurs ont estimé que les patients présentant de graves défauts OxPhos présenteraient de la même manière une efficacité métabolique altérée et une dépense énergétique au repos accrue, un état connu sous le nom d'hypermétabolisme. D'autres causes de défauts OxPhos, y compris des mutations dans les gènes nucléaires codant pour des facteurs d'assemblage de la chaîne respiratoire comme SURF127, qui provoquent des maladies et réduisent la durée de vie chez l'homme, pourraient également déclencher un hypermétabolisme.
Existe-t-il un lien de causalité entre l'hypermétabolisme et la durée de vie chez l'homme ? Chez les individus en bonne santé, un dépense énergétique au repos (REE) élevé ou un hypermétabolisme mesuré par calorimétrie indirecte (consommation d'oxygène, VO2) prédit un déclin physiologique lié à l'âge plus rapide et prédit indépendamment une mortalité de 25 à 53 % plus élevée au cours des 20 à 40 années suivantes.
Mécaniquement, de multiples processus se disputent des ressources énergétiques limitées au sein des cellules, ainsi qu'au sein des organismes, en particulier dans des conditions d'énergie restreinte. Certaines opérations cellulaires sont prioritaires sur d'autres. En conséquence, le coût énergétique des réponses au stress et leur augmentation associée de la transcription/traduction peuvent inhiber la croissance et la division cellulaire, voire déclencher une sénescence prématurée. Récemment, il a été rapporté qu'une activation excessive de l'ISR inhibe à elle seule la croissance de la population cellulaire. Ainsi, l'activation de l'ISR induite par OxPhos et l'hypermétabolisme qui en résulte pourraient freiner la croissance et/ou provoquer une mort prématurée en forçant un compromis énergétique entre les réponses au stress et les voies de croissance/survie.
En simplifiant le processus serait le suivant : (i) les défauts génétiques mitochondriaux d'OxPhos déclenchent des ISR, (ii) les cellules fonctionnent sous des contraintes énergétiques où la priorisation des réponses au stress et des coûts de transcription/traduction peuvent précipiter la sénescence, (iii) une diminution de l'efficacité métabolique prédit une durée de vie plus courte chez l'homme et d'autres animaux
Les auteurs ont d'abord testé cette hypothèse en analysant à nouveau les données de plusieurs cohortes cliniques de maladies mitochondriales primaires avec des évaluations directes et indirectes de la dépense énergétique et de la durée de vie, puis via des études longitudinales in vitro sur des fibroblastes primaires humains et dérivés de patients. Ils ont développé deux systèmes modèles cellulaires qui montrent que les défauts génétiques et pharmacologiques mitochondriaux d'OxPhos déclenchent un hypermétabolisme marqué d'une manière cellulaire autonome. Malgré le mode d'action divergent des modèles SURF1 et Oligo, ainsi que certaines réponses moléculaires divergentes, les deux modèles convergent sur le même phénotype hypermétabolique
Tout d'abord, les auteurs ont observé que le marqueur de maladie mitochondriale GDF15 était largement indétectable dans les milieux de jeunes fibroblastes sains, mais augmentait progressivement tout au long de la vie cellulaire. Cette découverte récapitule l'augmentation liée à l'âge du GDF15 chez l'homme, le GDF15 extracellulaire avait tendance à être élevé dans les deux modèles de défauts d'OxPhos.
Deuxièmement, les auteurs ont observé que le dysfonctionnement d'OxPhos dû aux mutations SURF1 et, dans une moindre mesure, au traitement Oligo, provoquaient tous deux une instabilité secondaire du génome mitochondrial. L'instabilité du génome mitochondrial était associée à l'accumulation variable de délétions du génome mitochondrial tout au long de la vie cellulaire, et non à cause de mutations ponctuelles.
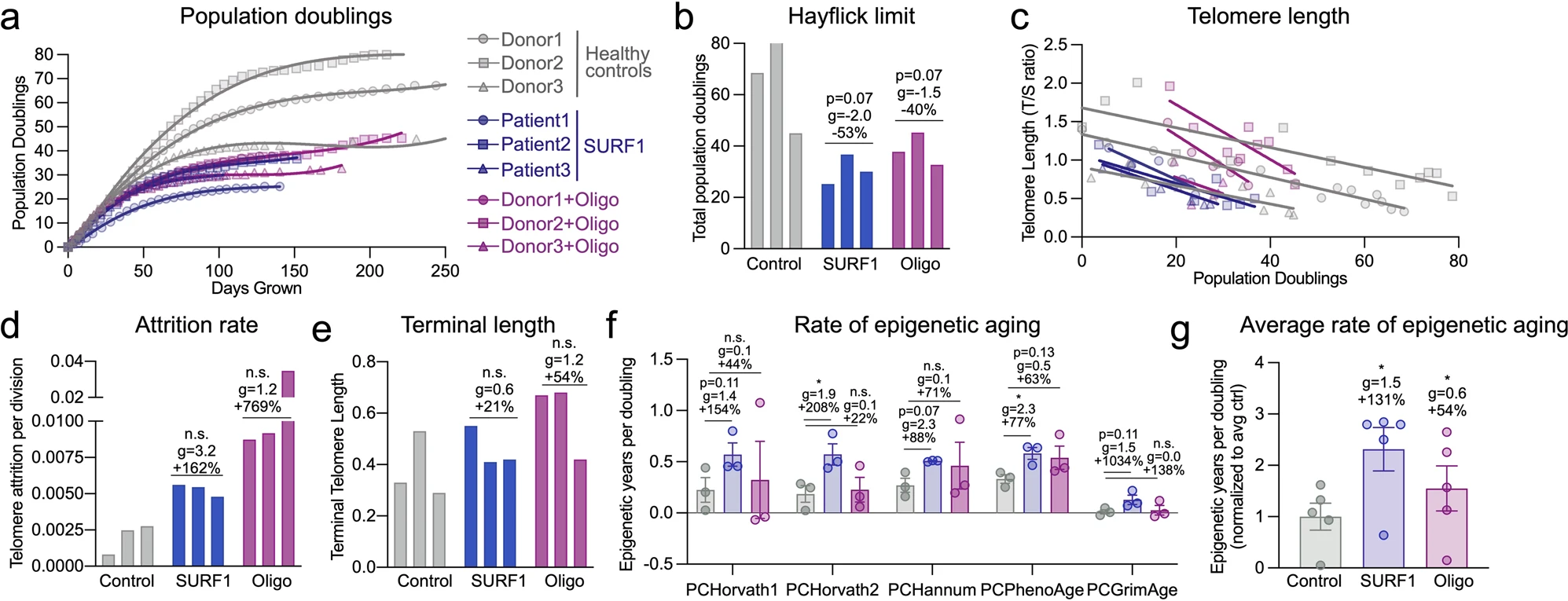 Troisièmement, les défauts mitochondriaux d'OxPhos ont considérablement augmenté le taux d'érosion des télomères par division cellulaire, malgré la régulation adaptative de la transcription des composants du complexe de protection des télomères. Une étude sur le muscle squelettique d'enfants présentant de fortes mutations hétéroplasmiques du génome mitochondrial a également rapporté des télomères excessivement courts, d'une longueur similaire aux télomères de témoins sains de 80 ans. Le compromis énergétique ou la « compétition » entre la traduction et la croissance pourrait expliquer pourquoi les cellules déficientes en OxPhos, qui doivent dépenser une grande partie de leur budget énergétique pour réguler à la hausse la transcription/traduction et la sécrétion, se développent également plus lentement. La réplication de l'ADN est également contrainte énergétiquement et se situe notamment au bas d'une hiérarchie de processus énergivores où les processus vitaux, c'est-à-dire que dans une situation où l'énergie est limitée, l'équilibre ionique et la traduction sont prioritaires sur la division et la réplication de l'ADN.
Troisièmement, les défauts mitochondriaux d'OxPhos ont considérablement augmenté le taux d'érosion des télomères par division cellulaire, malgré la régulation adaptative de la transcription des composants du complexe de protection des télomères. Une étude sur le muscle squelettique d'enfants présentant de fortes mutations hétéroplasmiques du génome mitochondrial a également rapporté des télomères excessivement courts, d'une longueur similaire aux télomères de témoins sains de 80 ans. Le compromis énergétique ou la « compétition » entre la traduction et la croissance pourrait expliquer pourquoi les cellules déficientes en OxPhos, qui doivent dépenser une grande partie de leur budget énergétique pour réguler à la hausse la transcription/traduction et la sécrétion, se développent également plus lentement. La réplication de l'ADN est également contrainte énergétiquement et se situe notamment au bas d'une hiérarchie de processus énergivores où les processus vitaux, c'est-à-dire que dans une situation où l'énergie est limitée, l'équilibre ionique et la traduction sont prioritaires sur la division et la réplication de l'ADN.
Quatrièmement, leurs ensembles de données longitudinaux RNASeq et DNAm révèlent des recalibrages conservés impliquant des voies de développement et de traduction, ainsi que la communication cellule-cellule, avec des défauts OxPhos et un hypermétabolisme. Le développement neuronal, la signalisation cellulaire, la morphogenèse, le cycle cellulaire et le métabolisme étaient les processus prédominants altérés dans les maladies liées à SURF1. L'induction de ces voies énergétiquement exigeantes qui limitent la croissance au niveau cellulaire et éventuellement au niveau de l'organisme, pourrait aider à expliquer pourquoi une caractéristique majeure des troubles mitochondriaux pédiatriques (y compris leurs donneurs SURF1) est un retard de développement neurologique, et aussi pourquoi les patients adultes présentent couramment petite taille.
Ainsi, l'organisme soumis à un stress métabolique n'initie pas un état hypométabolique d'économie d'énergie avec une activité de signalisation réduite, mais active à la place des réponses de stress énergivores (ISR), qui doivent détourner et consommer des ressources énergétiques, forçant ainsi un compromis apparent avec d'autres processus tels que la croissance. et les voies de longévité.
L'ATP peut être produit par un certain nombre de processus cellulaires distincts; les trois voies principales chez les eucaryotes sont la glycolyse, le cycle de l'acide citrique/phosphorylation oxydative et la bêta-oxydation. Dans les cellules eucaryotes, le cycle de l'acide citrique se produit dans la matrice de la mitochondrie. Les défauts OxPhos dans les fibroblastes ont déclenché une évolution vers la production d'ATP glycolytique. Le déplacement glycolytique est cohérent avec le déplacement physiologique de l'oxydation du substrat des lipides/acides aminés vers les glucides. Cela pourrait s'expliquer sur la base des contraintes énergétiques et de l'efficacité protéomique, puisque le coût protéomique d'OxPhos est au moins le double de celui de la fermentation glycolytique. Ainsi, les cellules peuvent "choisir" de détourner le flux métabolique vers la glycolyse même lorsque OxPhos est au moins partiellement fonctionnel, comme dans le cancer, en raison des contraintes énergétiques intracellulaires croissantes induites par l'hypermétabolisme.
L'alcool peut provoquer un hypermétabolisme chez les individus en bonne santé, augmentant les dépense énergétique au repos (REE) du corps entier jusqu'à 16 % et inhibant l'oxydation des lipides de 31 à 36 %. L'alcool peut donc aggraver un hypermétabolisme préexistant, imposant ainsi des contraintes énergétiques supplémentaires sur les fonctions cellulaires ou physiologiques vitales.
L'hypermétabolisme chronique pourrait, en partie, expliquer pourquoi les infections peuvent déclencher des exacerbations cliniques. Le coût métabolique de l'activation immunitaire contre les infections virales et bactériennes est élevé et la production de cytokines dans les leucocytes humains est sous régulation mitochondriale. Ainsi, l'immunité doit donc concurrencer les autres systèmes de maintenance de l'hôte. Nous supposons que dans les maladies mitochondriales, parce que les ressources énergétiques limitées sont consommées à un rythme plus élevé que la normale en raison de l'hypermétabolisme systémique, les patients peuvent manquer de la réserve énergétique nécessaire pour maintenir les organes vitaux tout en développant des réponses immunitaires adéquates.
Une question ouverte majeure concerne l'origine et la possibilité de modifier les voies de signalisation et les processus cellulaires qui sous-tendent l'hypermétabolisme dans les cellules et les humains déficients en OxPhos. Plutôt que de poursuivre une seule explication potentielle, les auteurs ont tenté ici de phénotyper profondément les deux modèles cellulaires d'hypermétabolisme et de produire un ensemble de données de base couvrant plusieurs processus et voies clés précédemment impliqués dans la pathogenèse des défauts d'OxPhos chez les humains et les modèles animaux. Leur ensemble de données fournit donc une base qui peut être utilisée comme ressource pour développer des expériences mécanistes ciblées pour (i) déterminer l'origine et la possibilité de modification de l'hypermétabolisme dans le contexte des défauts OxPhos in vitro et in vivo, et (ii) résoudre le mécanisme(s) reliant l'hypermétabolisme à la biologie du vieillissement humain.
En conclusion, ces données translationnelles fournissent donc une base pour rationaliser certaines caractéristiques cliniques inexpliquées des maladies mitochondriales et suggèrent que les compromis énergétiques intracellulaires et systémiques (plutôt que le déficit en ATP) peuvent contribuer à la pathogenèse des maladies mitochondriales. Le cadre explicatif proposé de l'hypermétabolisme cellulaire et physiologique appelle des études bien contrôlées pour mieux comprendre dans quelle mesure l'hypermétabolisme est un spectateur ou un signe avant-coureur de la morbidité et de la mortalité précoce chez les patients atteints de maladies mitochondriales. Leurs résultats translationnels mettent en évidence la nécessité de partenariats collaboratifs qui relient les aspects cellulaires, cliniques et signalés par les patients des maladies mitochondriales et du vieillissement. J'y ajouterais les patients atteints de maladies neurodégénaratives.