Inhibiting cGAS-STING pathway confers resilience against Alzheimer's . '
Blood Lipoprotein Levels and Alzheimer Disease . '
Certains médicaments contre le VIH et l’hépatite pourraient contribuer à... . '
Perineuronal net modulation in a Parkinson's disease mouse model . '
Parkinson's disease and acupuncture . '
How to cope with the deluge of scientific publication? . '
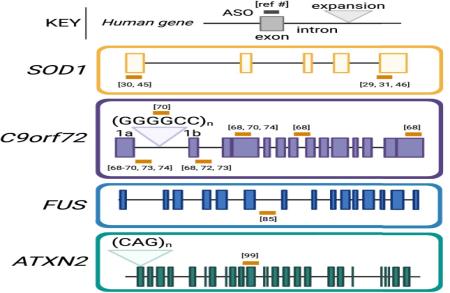
Jacifusen pour FUS-ALS : une étude de cas . '
Small molecule may dissolve stress granules . '
Age-dependent accumulation of mitochondrial tRNA mutations in mouse kidneys linked to mitochondrial kidney diseases. . '
A reference model of circulating hematopoietic stem cells across the lifespan with applications to diagnostics. . '
DNA methylation profiles of long-term cannabis users in midlife: a comprehensive evaluation of published... . '
Movement-responsive deep brain stimulation for Parkinson's disease using a remotely optimized neural decoder. . '
Plasma lipidomic fingerprinting enables high-accuracy biomarker discovery for Alzheimer's disease: a targeted LC-MRM/MS... . '