For years, the discourse surrounding familial amyotrophic lateral sclerosis (ALS) has largely focused on genomic instability, yet we know that there is a significant metastatic aspect to ALS, if only through the observed loss of muscle mass.
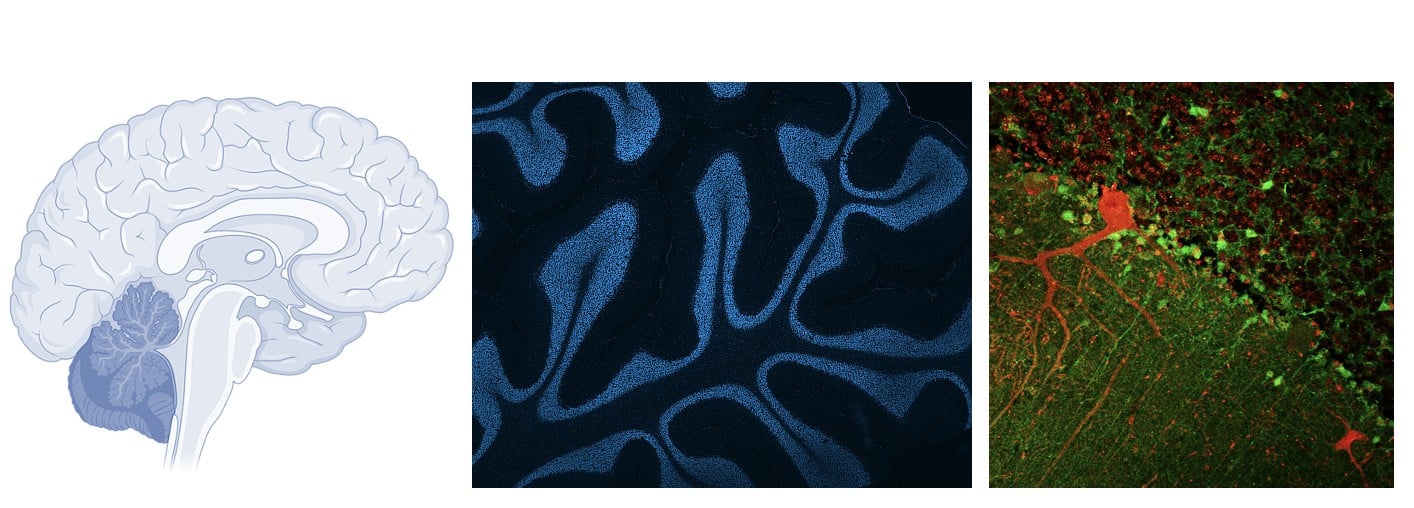
Rather than focusing solely on the motor regions of the brain, researchers have turned their attention to the cerebellum, a brain region associated with balance and coordination, but long underestimated in ALS. It is unfortunately a minor theme in research that ALS is not a specific impairment of motor neurons, as the controversial Dr. Charcot believed more than 150 years ago.
It has long been known that, in the brains of patients with the most common genetic variant of the disease, a mutation in the C9orf72 gene, neurons accumulate catastrophic double-strand breaks in DNA. The neurons then generate abnormal proteins from this corrupted DNA. Yet, a crucial piece of the puzzle was missing.
Previously, scientists observed these structural lesions but didn't know why the cells suddenly lost the ability to repair their own DNA. A study published in the journal Brain (Singh et al., 2026) identified a biochemical "intermediary" that fills this gap: an enzyme called PAICS. These findings suggest that the neurodegeneration associated with this form of ALS could be fundamentally due to a shortage of cellular energy.
In the most common genetic form of the disease (linked to the C9orf72 gene) in zebrafish, Singh et al. observed early cerebellar atrophy, characterized by the loss of two essential types of neurons: Purkinje cells and granule cells, well before the onset of motor symptoms.
 To understand this major breakthrough, it's helpful to dispel a common misconception about DNA damage. Our genetic code isn't a static library; it constantly undergoes routine damage. Every time a brain cell processes oxygen, generates metabolic energy, or emits an electrical impulse, it creates oxidative stress that naturally breaks its own DNA strands.
To understand this major breakthrough, it's helpful to dispel a common misconception about DNA damage. Our genetic code isn't a static library; it constantly undergoes routine damage. Every time a brain cell processes oxygen, generates metabolic energy, or emits an electrical impulse, it creates oxidative stress that naturally breaks its own DNA strands.
In a healthy neuron, efficient, automated maintenance systems quickly repair these breaks using nucleotides. The cell uses two pathways to acquire these nucleotides: recycling existing materials or synthesizing them de novo.
In ALS linked to the C9orf72 gene, this maintenance loop malfunctions. Although researchers were able to observe the resulting genomic breaks, the underlying cause of the repair mechanism's failure remained unknown.
Discovering the bottleneck: a purine shortage. The C9orf72 mutation is characterized by a massive, repetitive expansion of the genetic code ($GGGGCC$). This expansion forces the cell to produce abnormal, sticky protein chains called dipeptide repeat proteins (DPRs). These toxic DPRs have been shown to act as cellular disruptors, specifically inhibiting the expression of the PAICS gene.
[Hereditary C9orf72 Mutation]
▼
[Generation of Toxic DPR Proteins]
▼
[Suppression of PAICS Enzyme Production] ◄── The Missing Link
▼
[Purine Shortage (Lack of Precursors for Repair)]
▼
[Unrepaired DNA Breaks and Ultimately, Neuronal Death]
The PAICS gene codes for an enzyme essential for the de novo synthesis of purines, the essential amino acids (adenine and guanine) that make up our DNA.
When DPR suppresses PAICS expression, the neuron enters a state of severe purine deficiency. The DNA damage caused by daily brain activity remains unchanged. What changes is that the cell is completely deprived of the elements necessary for its repair. Without purines, repair mechanisms break down, DNA abnormalities accumulate, and the neuron eventually degenerates.
Beyond the sensationalism: what are the implications for therapies? When laboratory breakthroughs like this one occur, institutional press releases often claim that a cure to “stop ALS” is imminent. It is essential to temper these expectations with the realities of translational medicine. Researchers have succeeded in reversing this purine deficiency and saving neurons in human cell cultures in the laboratory and in animal models (zebrafish). However, translating these results into human therapies represents a significant challenge.
While scientists have validated this biological target, they have not yet developed a safe and stable drug capable of exploiting the human blood-brain barrier, an approved treatment based on this approach is unlikely to be available for another 8 to 12 years, or even longer. In addition a major challenge remains: how to selectively upregulate purine biosynthesis enzymes in the CNS without promoting oncogenic pathways, given that PAICS is also known to be overexpressed in various cancers?
Despite this long delay, the discovery is structurally important for two reasons: For patients who inherit the C9orf72 mutation, a future drug that artificially upregulates PAICS could act as a protective shield. This would not erase the underlying genetic mutation, but it could halt the resulting purine deficiency, thus preserving neurons.
Cross-applicability to other ALS subtypes: While this specific PAICS bottleneck is directly triggered by C9orf72-type genetic abnormalities, independent research shows that other aggressive forms of ALS, such as those involving TDP-43 and FUS mutations, also converge toward a fatal inability to repair DNA. If we can develop a method to manipulate PAICS to flood diseased neurons with an excess of purines, the building blocks of the disease, we could effectively stimulate alternative cellular repair mechanisms.
From a clinical perspective, the observation that cerebellar changes precede motor symptoms is particularly interesting. Cerebellar imaging or biomarkers related to purine metabolism could potentially serve as early diagnostic tools.
By shifting the focus from the structural lesions themselves to the metabolic pathways that repair them, this research offers a concrete and elegant framework for future neuroprotective therapies.

 By testing this approach on mouse models of both diseases and on human patient stem cells (iPSCs), the team observed several crucial improvements:
By testing this approach on mouse models of both diseases and on human patient stem cells (iPSCs), the team observed several crucial improvements: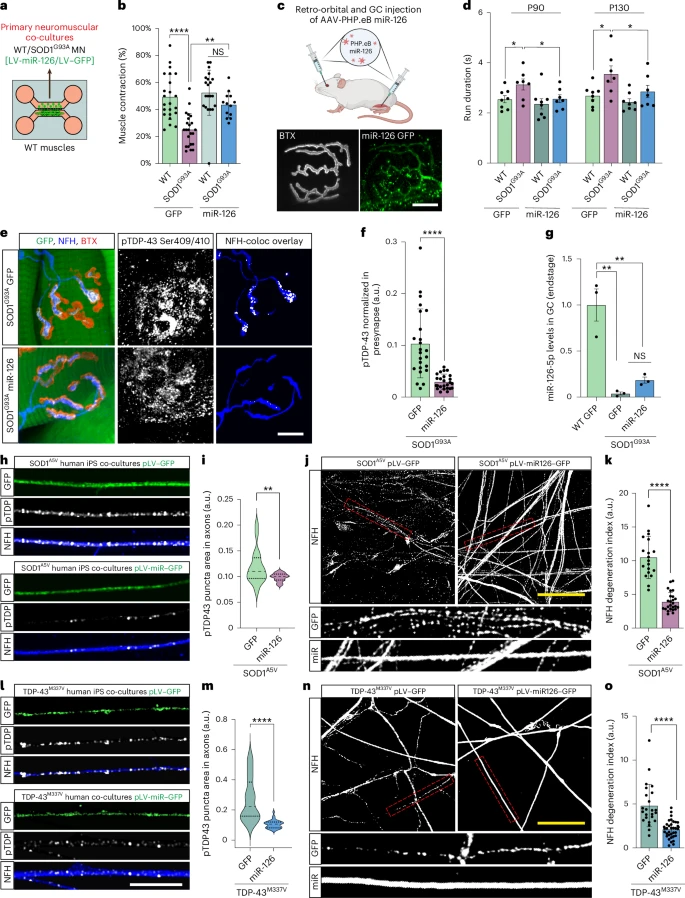 When pathologists examine the spinal motor neurons of patients with SOD1-related ALS, the nuclei generally appear normal: the TDP-43 protein is always present, and abnormal aggregates are rarely observed. This is why SOD1-related ALS has been considered "TDP-43 negative."
When pathologists examine the spinal motor neurons of patients with SOD1-related ALS, the nuclei generally appear normal: the TDP-43 protein is always present, and abnormal aggregates are rarely observed. This is why SOD1-related ALS has been considered "TDP-43 negative."